PART 493 - LABORATORY REQUIREMENTS
Source:
55 FR 9576, Mar. 14, 1990, unless otherwise noted.
Subpart A - General Provisions
Source:
57 FR 7139, Feb. 28, 1992, unless otherwise noted.
§ 493.1 Basis and scope.
This part sets forth the conditions that all laboratories must meet to be certified to perform testing on human specimens under the Clinical Laboratory Improvement Amendments of 1988 (CLIA). It implements sections 1861(e) and (j), the sentence following section 1861(s)(13), and 1902(a)(9) of the Social Security Act, and section 353 of the Public Health Service Act, as amended by section 2 of the Taking Essential Steps for Testing Act of 2012. This part applies to all laboratories as defined under “laboratory” in § 493.2 of this part. This part also applies to laboratories seeking payment under the Medicare and Medicaid programs. The requirements are the same for Medicare approval as for CLIA certification.
[57 FR 7139, Feb. 28, 1992, as amended at 79 FR 25480, May 2, 2014]
§ 493.2 Definitions.
As used in this part, unless the context indicates otherwise -
Accredited institution means a school or program which -
(a) Admits as regular student only persons having a certificate of graduation from a school providing secondary education, or the recognized equivalent of such certificate;
(b) Is legally authorized within the State to provide a program of education beyond secondary education;
(c) Provides an educational program for which it awards a bachelor's degree or provides not less than a 2-year program which is acceptable toward such a degree, or provides an educational program for which it awards a master's or doctoral degree;
(d) Is accredited by a nationally recognized accrediting agency or association.
This definition includes any foreign institution of higher education that HHS or its designee determines meets substantially equivalent requirements.
Accredited laboratory means a laboratory that has voluntarily applied for and been accredited by a private, nonprofit accreditation organization approved by CMS in accordance with this part;
Adverse action means the imposition of a principal or alternative sanction by CMS.
ALJ stands for Administrative Law Judge.
Alternative sanctions means sanctions that may be imposed in lieu of or in addition to principal sanctions. The term is synonymous with “intermediate sanctions” as used in section 1846 of the Act.
Analyte means a substance or constituent for which the laboratory conducts testing.
Approved accreditation organization for laboratories means a private, nonprofit accreditation organization that has formally applied for and received CMS's approval based on the organization's compliance with this part.
Approved State laboratory program means a licensure or other regulatory program for laboratories in a State, the requirements of which are imposed under State law, and the State laboratory program has received CMS approval based on the State's compliance with this part.
Authorized person means an individual authorized under State law to order tests or receive test results, or both.
Calibration means a process of testing and adjusting an instrument or test system to establish a correlation between the measurement response and the concentration or amount of the substance that is being measured by the test procedure.
Calibration verification means the assaying of materials of known concentration in the same manner as patient samples to substantiate the instrument or test system's calibration throughout the reportable range for patient test results.
Challenge means, for quantitative tests, an assessment of the amount of substance or analyte present or measured in a sample. For qualitative tests, a challenge means the determination of the presence or the absence of an analyte, organism, or substance in a sample.
CLIA means the Clinical Laboratory Improvement Amendments of 1988.
CLIA certificate means any of the following types of certificates issued by CMS or its agent:
(1) Certificate of compliance means a certificate issued to a laboratory after an inspection that finds the laboratory to be in compliance with all applicable condition level requirements, or reissued before the expiration date, pending an appeal, in accordance with § 493.49, when an inspection has found the laboratory to be out of compliance with one or more condition level requirements.
(2) Certificate for provider-performed microscopy (PPM) procedures means a certificate issued or reissued before the expiration date, pending an appeal, in accordance with § 493.47, to a laboratory in which a physician, midlevel practitioner or dentist performs no tests other than PPM procedures and, if desired, waived tests listed in § 493.15(c).
(3) Certificate of accreditation means a certificate issued on the basis of the laboratory's accreditation by an accreditation organization approved by CMS (indicating that the laboratory is deemed to meet applicable CLIA requirements) or reissued before the expiration date, pending an appeal, in accordance with § 493.61, when a validation or complaint survey has found the laboratory to be noncompliant with one or more CLIA conditions.
(4) Certificate of registration or registration certificate means a certificate issued or reissued before the expiration date, pending an appeal, in accordance with § 493.45, that enables the entity to conduct moderate or high complexity laboratory testing or both until the entity is determined to be in compliance through a survey by CMS or its agent; or in accordance with § 493.57 to an entity that is accredited by an approved accreditation organization.
(5) Certificate of waiver means a certificate issued or reissued before the expiration date, pending an appeal, in accordance with § 493.37, to a laboratory to perform only the waived tests listed at § 493.15(c).
CLIA-exempt laboratory means a laboratory that has been licensed or approved by a State where CMS has determined that the State has enacted laws relating to laboratory requirements that are equal to or more stringent than CLIA requirements and the State licensure program has been approved by CMS in accordance with subpart E of this part.
Condition level deficiency means noncompliance with one or more condition level requirements.
Condition level requirements means any of the requirements identified as “conditions” in § 493.41 and subparts G through Q of this part.
Confirmatory testing means testing performed by a second analytical procedure that could be used to substantiate or bring into question the result of an initial laboratory test.
Credible allegation of compliance means a statement or documentation that -
(1) Is made by a representative of a laboratory that has a history of having maintained a commitment to compliance and of taking corrective action when required;
(2) Is realistic in terms of its being possible to accomplish the required corrective action between the date of the exit conference and the date of the allegation; and
(3) Indicates that the problem has been resolved.
Dentist means a doctor of dental medicine or doctor of dental surgery licensed by the State to practice dentistry within the State in which the laboratory is located.
Distributive testing means laboratory testing performed on the same specimen, or an aliquot of it, that requires sharing it between two or more laboratories to obtain all data required to complete an interpretation or calculation necessary to provide a final reportable result for the originally ordered test. When such testing occurs at multiple locations with different CLIA certificates, it is considered distributive testing.
Equivalency means that an accreditation organization's or a State laboratory program's requirements, taken as a whole, are equal to or more stringent than the CLIA requirements established by CMS, taken as whole. It is acceptable for an accreditation organization's or State laboratory program's requirements to be organized differently or otherwise vary from the CLIA requirements, as long as
(1) all of the requirements taken as a whole would provide at least the same protection as the CLIA requirements taken as a whole; and
(2) a finding of noncompliance with respect to CLIA requirements taken as a whole would be matched by a finding of noncompliance with the accreditation or State requirements taken as a whole.
CMS agent means an entity with which CMS arranges to inspect laboratories and assess laboratory activities against CLIA requirements and may be a State survey agency, a private, nonprofit organization other than an approved accreditation organization, a component of HHS, or any other governmental component CMS approves for this purpose. In those instances where all of the laboratories in a State are exempt from CLIA requirements, based on the approval of a State's exemption request, the State survey agency is not the CMS agent.
FDA-cleared or approved test system means a test system cleared or approved by the FDA through the premarket notification (510(k)) or premarket approval (PMA) process for in-vitro diagnostic use. Unless otherwise stated, this includes test systems exempt from FDA premarket clearance or approval.
HHS means the Department of Health and Human Services, or its designee.
Immediate jeopardy means a situation in which immediate corrective action is necessary because the laboratory's noncompliance with one or more condition level requirements has already caused, is causing, or is likely to cause, at any time, serious injury or harm, or death, to individuals served by the laboratory or to the health or safety of the general public. This term is synonymous with imminent and serious risk to human health and significant hazard to the public health.
Intentional violation means knowing and willful noncompliance with any CLIA condition.
Kit means all components of a test that are packaged together.
Laboratory means a facility for the biological, microbiological, serological, chemical, immunohematological, hematological, biophysical, cytological, pathological, or other examination of materials derived from the human body for the purpose of providing information for the diagnosis, prevention, or treatment of any disease or impairment of, or the assessment of the health of, human beings. These examinations also include procedures to determine, measure, or otherwise describe the presence or absence of various substances or organisms in the body. Facilities only collecting or preparing specimens (or both) or only serving as a mailing service and not performing testing are not considered laboratories.
Midlevel practitioner means a nurse midwife, nurse practitioner, or physician assistant, licensed by the State within which the individual practices, if such licensing is required in the State in which the laboratory is located.
Nonwaived test means any test system, assay, or examination that has not been found to meet the statutory criteria specified at section 353(d)(3) of the Public Health Service Act.
Operator means the individual or group of individuals who oversee all facets of the operation of a laboratory and who bear primary responsibility for the safety and reliability of the results of all specimen testing performed in that laboratory. The term includes -
(1) A director of the laboratory if he or she meets the stated criteria; and
(2) The members of the board of directors and the officers of a laboratory that is a small corporation under subchapter S of the Internal Revenue Code.
Owner means any person who owns any interest in a laboratory except for an interest in a laboratory whose stock and/or securities are publicly traded. (That is e.g., the purchase of shares of stock or securities on the New York Stock Exchange in a corporation owning a laboratory would not make a person an owner for the purpose of this regulation.)
Party means a laboratory affected by any of the enforcement procedures set forth in this subpart, by CMS or the OIG, as appropriate.
Performance characteristic means a property of a test that is used to describe its quality, e.g., accuracy, precision, analytical sensitivity, analytical specificity, reportable range, reference range, etc.
Performance specification means a value or range of values for a performance characteristic, established or verified by the laboratory, that is used to describe the quality of patient test results.
Physician means an individual with a doctor of medicine, doctor of osteopathy, or doctor of podiatric medicine degree who is licensed by the State to practice medicine, osteopathy, or podiatry within the State in which the laboratory is located.
Principal sanction means the suspension, limitation, or revocation of any type of CLIA certificate or the cancellation of the laboratory's approval to receive Medicare payment for its services.
Prospective laboratory means a laboratory that is operating under a registration certificate or is seeking any of the three other types of CLIA certificates.
Rate of disparity means the percentage of sample validation inspections for a specific accreditation organization or State where CMS, the State survey agency or other CMS agent finds noncompliance with one or more condition level requirements but no comparable deficiencies were cited by the accreditation organization or the State, and it is reasonable to conclude that the deficiencies were present at the time of the most recent accreditation organization or State licensure inspection.
Example:
Assume the State survey agency, CMS or other CMS agent performs 200 sample validation inspections for laboratories accredited by a single accreditation organization or licensed in an exempt State during a validation review period and finds that 60 of the 200 laboratories had one or more condition level requirements out of compliance. CMS reviews the validation and accreditation organization's or State's inspections of the validated laboratories and determines that the State or accreditation organization found comparable deficiencies in 22 of the 60 laboratories and it is reasonable to conclude that deficiencies were present in the remaining 38 laboratories at the time of the accreditation organization's or State's inspection. Thirty-eight divided by 200 equals a 19 percent rate of disparity.
Referee laboratory means a laboratory currently in compliance with applicable CLIA requirements, that has had a record of satisfactory proficiency testing performance for all testing events for at least one year for a specific test, analyte, subspecialty, or specialty and has been designated by an HHS approved proficiency testing program as a referee laboratory for analyzing proficiency testing specimens for the purpose of determining the correct response for the specimens in a testing event for that specific test, analyte, subspecialty, or specialty.
Reference range means the range of test values expected for a designated population of individuals, e.g., 95 percent of individuals that are presumed to be healthy (or normal).
Reflex testing means confirmatory or additional laboratory testing that is automatically requested by a laboratory under its standard operating procedures for patient specimens when the laboratory's findings indicate test results that are abnormal, are outside a predetermined range, or meet other pre-established criteria for additional testing.
Repeat proficiency testing referral means a second instance in which a proficiency testing sample, or a portion of a sample, is referred, for any reason, to another laboratory for analysis prior to the laboratory's proficiency testing program event cut-off date within the period of time encompassing the two prior survey cycles (including initial certification, recertification, or the equivalent for laboratories surveyed by an approved accreditation organization).
Reportable range means the span of test result values over which the laboratory can establish or verify the accuracy of the instrument or test system measurement response.
Sample in proficiency testing means the material contained in a vial, on a slide, or other unit that contains material to be tested by proficiency testing program participants. When possible, samples are of human origin.
State includes, for purposes of this part, each of the 50 States, the District of Columbia, the Commonwealth of Puerto Rico, the Virgin Islands and a political subdivision of a State where the State, acting pursuant to State law, has expressly delegated powers to the political subdivision sufficient to authorize the political subdivision to act for the State in enforcing requirements equal to or more stringent than CLIA requirements.
State licensure means the issuance of a license to, or the approval of, a laboratory by a State laboratory program as meeting standards for licensing or approval established under State law.
State licensure program means a State laboratory licensure or approval program.
State survey agency means the State health agency or other appropriate State or local agency that has an agreement under section 1864 of the Social Security Act and is used by CMS to perform surveys and inspections.
Substantial allegation of noncompliance means a complaint from any of a variety of sources (including complaints submitted in person, by telephone, through written correspondence, or in newspaper or magazine articles) that, if substantiated, would have an impact on the health and safety of the general public or of individuals served by a laboratory and raises doubts as to a laboratory's compliance with any condition level requirement.
Target value for quantitative tests means either the mean of all participant responses after removal of outliers (those responses greater than 3 standard deviations from the original mean) or the mean established by definitive or reference methods acceptable for use in the National Reference System for the Clinical Laboratory (NRSCL) by the National Committee for the Clinical Laboratory Standards (NCCLS). In instances where definitive or reference methods are not available or a specific method's results demonstrate bias that is not observed with actual patient specimens, as determined by a defensible scientific protocol, a comparative method or a method group (“peer” group) may be used. If the method group is less than 10 participants, “target value” means the overall mean after outlier removal (as defined above) unless acceptable scientific reasons are available to indicate that such an evaluation is not appropriate.
Test system means the instructions and all of the instrumentation, equipment, reagents, and supplies needed to perform an assay or examination and generate test results.
Unsatisfactory proficiency testing performance means failure to attain the minimum satisfactory score for an analyte, test, subspecialty, or specialty for a testing event.
Unsuccessful participation in proficiency testing means any of the following:
(1) Unsatisfactory performance for the same analyte in two consecutive or two out of three testing events.
(2) Repeated unsatisfactory overall testing event scores for two consecutive or two out of three testing events for the same specialty or subspecialty.
(3) An unsatisfactory testing event score for those subspecialties not graded by analyte (that is, bacteriology, mycobacteriology, virology, parasitology, mycology, blood compatibility, immunohematology, or syphilis serology) for the same subspecialty for two consecutive or two out of three testing events.
(4) Failure of a laboratory performing gynecologic cytology to meet the standard at § 493.855.
Unsuccessful proficiency testing performance means a failure to attain the minimum satisfactory score for an analyte, test, subspecialty, or specialty for two consecutive or two of three consecutive testing events.
Validation review period means the one year time period during which CMS conducts validation inspections and evaluates the results of the most recent surveys performed by an accreditation organization or State laboratory program.
Waived test means a test system, assay, or examination that HHS has determined meets the CLIA statutory criteria as specified for waiver under section 353(d)(3) of the Public Health Service Act.
[57 FR 7139, Feb. 28, 1992, as amended at 57 FR 7236, Feb. 28, 1992; 57 FR 34013, July 31, 1992; 57 FR 35761, Aug. 11, 1992; 58 FR 5220, Jan. 19, 1993; 58 FR 48323, Sept. 15, 1993; 60 FR 20043, Apr. 24, 1995; 63 FR 26732, May 14, 1998; 68 FR 3702, Jan. 24, 2003; 68 FR 50723, Aug. 22, 2003; 79 FR 25480, May 2, 2014; 79 FR 27157, May 12, 2014; 85 FR 54873, Sept. 2, 2020]
§ 493.3 Applicability.
(a) Basic rule. Except as specified in paragraph (b) of this section, a laboratory will be cited as out of compliance with section 353 of the Public Health Service Act unless it—
(1) Has a current, unrevoked or unsuspended certificate of waiver, registration certificate, certificate of compliance, certificate for PPM procedures, or certificate of accreditation issued by HHS applicable to the category of examinations or procedures performed by the laboratory; or
(2) Is CLIA-exempt.
(b) Exception. These rules do not apply to components or functions of—
(1) Any facility or component of a facility that only performs testing for forensic purposes;
(2) Research laboratories that test human specimens but do not report patient specific results for the diagnosis, prevention or treatment of any disease or impairment of, or the assessment of the health of individual patients; or
(3) Laboratories certified by the Substance Abuse and Mental Health Services Administration (SAMHSA), in which drug testing is performed which meets SAMHSA guidelines and regulations. However, all other testing conducted by a SAMHSA-certified laboratory is subject to this rule.
(c) Federal laboratories. Laboratories under the jurisdiction of an agency of the Federal Government are subject to the rules of this part, except that the Secretary may modify the application of such requirements as appropriate.
[57 FR 7139, Feb. 28, 1992, as amended at 58 FR 5221, Jan. 19, 1993; 60 FR 20043, Apr. 24, 1995; 68 FR 3702, Jan. 24, 2003]
§ 493.5 Categories of tests by complexity.
(a) Laboratory tests are categorized as one of the following:
(1) Waived tests.
(2) Tests of moderate complexity, including the subcategory of PPM procedures.
(3) Tests of high complexity.
(b) A laboratory may perform only waived tests, only tests of moderate complexity, only PPM procedures, only tests of high complexity or any combination of these tests.
(c) Each laboratory must be either CLIA-exempt or possess one of the following CLIA certificates, as defined in § 493.2:
(1) Certificate of registration or registration certificate.
(2) Certificate of waiver.
(3) Certificate for PPM procedures.
(4) Certificate of compliance.
(5) Certificate of accreditation.
[60 FR 20043, Apr. 24, 1995]
§ 493.15 Laboratories performing waived tests.
(a) Requirement. Tests for certificate of waiver must meet the descriptive criteria specified in paragraph (b) of this section.
(b) Criteria. Test systems are simple laboratory examinations and procedures which—
(1) Are cleared by FDA for home use;
(2) Employ methodologies that are so simple and accurate as to render the likelihood of erroneous results negligible; or
(3) Pose no reasonable risk of harm to the patient if the test is performed incorrectly.
(c) Certificate of waiver tests. A laboratory may qualify for a certificate of waiver under section 353 of the PHS Act if it restricts the tests that it performs to one or more of the following tests or examinations (or additional tests added to this list as provided under paragraph (d) of this section) and no others:
(1) Dipstick or Tablet Reagent Urinalysis (non-automated) for the following:
(i) Bilirubin;
(ii) Glucose;
(iii) Hemoglobin;
(iv) Ketone;
(v) Leukocytes;
(vi) Nitrite;
(vii) pH;
(viii) Protein;
(ix) Specific gravity; and
(x) Urobilinogen.
(2) Fecal occult blood-non-automated;
(3) Ovulation tests—visual color comparison tests for human luteinizing hormone;
(4) Urine pregnancy tests—visual color comparison tests;
(5) Erythrocyte sedimentation rate—non-automated;
(6) Hemoglobin—copper sulfate—non-automated;
(7) Blood glucose by glucose monitoring devices cleared by the FDA specifically for home use;
(8) Spun microhematocrit; and
(9) Hemoglobin by single analyte instruments with self-contained or component features to perform specimen/reagent interaction, providing direct measurement and readout.
(d) Revisions to criteria for test categorization and the list of waived tests. HHS will determine whether a laboratory test meets the criteria listed under paragraph (b) of this section for a waived test. Revisions to the list of waived tests approved by HHS will be published in the Federal Register in a notice with opportunity for comment.
(e) Laboratories eligible for a certificate of waiver must—
(1) Follow manufacturers' instructions for performing the test; and
(2) Meet the requirements in subpart B, Certificate of Waiver, of this part.
[57 FR 7139, Feb. 28, 1992, as amended at 58 FR 5221, Jan. 19, 1993; 82 FR 48773, Oct. 20, 2017]
§ 493.17 Test categorization.
(a) Categorization by criteria. Notices will be published in the Federal Register which list each specific test system, assay, and examination categorized by complexity. Using the seven criteria specified in this paragraph for categorizing tests of moderate or high complexity, each specific laboratory test system, assay, and examination will be graded for level of complexity by assigning scores of 1, 2, or 3 within each criteria. The score of “1” indicates the lowest level of complexity, and the score of “3” indicates the highest level. These scores will be totaled. Test systems, assays or examinations receiving scores of 12 or less will be categorized as moderate complexity, while those receiving scores above 12 will be categorized as high complexity.
A score of “2” will be assigned to a criteria heading when the characteristics for a particular test are intermediate between the descriptions listed for scores of “1” and “3.”
(1) Knowledge —
(i) Score 1.
(A) Minimal scientific and technical knowledge is required to perform the test; and
(B) Knowledge required to perform the test may be obtained through on-the-job instruction.
(ii) Score 3. Specialized scientific and technical knowledge is essential to perform preanalytic, analytic or postanalytic phases of the testing.
(2) Training and experience —
(i) Score 1.
(A) Minimal training is required for preanalytic, analytic and postanalytic phases of the testing process; and
(B) Limited experience is required to perform the test.
(ii) Score 3.
(A) Specialized training is essential to perform the preanalytic, analytic or postanalytic testing process; or
(B) Substantial experience may be necessary for analytic test performance.
(3) Reagents and materials preparation —
(i) Score 1.
(A) Reagents and materials are generally stable and reliable; and
(B) Reagents and materials are prepackaged, or premeasured, or require no special handling, precautions or storage conditions.
(ii) Score 3.
(A) Reagents and materials may be labile and may require special handling to assure reliability; or
(B) Reagents and materials preparation may include manual steps such as gravimetric or volumetric measurements.
(4) Characteristics of operational steps —
(i) Score 1. Operational steps are either automatically executed (such as pipetting, temperature monitoring, or timing of steps), or are easily controlled.
(ii) Score 3. Operational steps in the testing process require close monitoring or control, and may require special specimen preparation, precise temperature control or timing of procedural steps, accurate pipetting, or extensive calculations.
(5) Calibration, quality control, and proficiency testing materials —
(i) Score 1.
(A) Calibration materials are stable and readily available;
(B) Quality control materials are stable and readily available; and
(C) External proficiency testing materials, when available, are stable.
(ii) Score 3.
(A) Calibration materials, if available, may be labile;
(B) Quality control materials may be labile, or not available; or
(C) External proficiency testing materials, if available, may be labile.
(6) Test system troubleshooting and equipment maintenance —
(i) Score 1.
(A) Test system troubleshooting is automatic or self-correcting, or clearly described or requires minimal judgment; and
(B) Equipment maintenance is provided by the manufacturer, is seldom needed, or can easily be performed.
(ii) Score 3.
(A) Troubleshooting is not automatic and requires decision-making and direct intervention to resolve most problems; or
(B) Maintenance requires special knowledge, skills, and abilities.
(7) Interpretation and judgment —
(i) Score 1.
(A) Minimal interpretation and judgment are required to perform preanalytic, analytic and postanalytic processes; and
(B) Resolution of problems requires limited independent interpretation and judgment; and
(ii) Score 3.
(A) Extensive independent interpretation and judgment are required to perform the preanalytic, analytic or postanalytic processes; and
(B) Resolution of problems requires extensive interpretation and judgment.
(b) Revisions to the criteria for categorization. The Clinical Laboratory Improvement Advisory Committee, as defined in subpart T of this part, will conduct reviews upon request of HHS and recommend to HHS revisions to the criteria for categorization of tests.
(c) Process for device/test categorization utilizing the scoring system under § 493.17(a).
(1)
(i) For new commercial test systems, assays, or examinations, the manufacturer, as part of its 510(k) and PMA application to FDA, will submit supporting data for device/test categorization. FDA will determine the complexity category, notify the manufacturers directly, and will simultaneously inform both CMS and CDC of the device/test category. FDA will consult with CDC concerning test categorization in the following three situations:
(A) When categorizing previously uncategorized new technology;
(B) When FDA determines it to be necessary in cases involving a request for a change in categorization; and
(C) If a manufacturer requests review of a categorization decision by FDA in accordance with 21 CFR 10.75.
(ii) Test categorization will be effective as of the notification to the applicant.
(2) For test systems, assays, or examinations not commercially available, a laboratory or professional group may submit a written request for categorization to PHS. These requests will be forwarded to CDC for evaluation; CDC will determine complexity category and notify the applicant, CMS, and FDA of the categorization decision. In the case of request for a change of category or for previously uncategorized new technology, PHS will receive the request application and forward it to CDC for categorization.
(3) A request for recategorization will be accepted for review if it is based on new information not previously submitted in a request for categorization or recategorization by the same applicant and will not be considered more frequently than once per year.
(4) If a laboratory test system, assay or examination does not appear on the lists of tests in the Federal Register notices, it is considered to be a test of high complexity until PHS, upon request, reviews the matter and notifies the applicant of its decision. Test categorization is effective as of the notification to the applicant.
(5) PHS will publish revisions periodically to the list of moderate and high complexity tests in the Federal Register in a notice with opportunity for comment.
[57 FR 7139, Feb. 28, 1992, as amended at 58 FR 5222, Jan. 19, 1993]
§ 493.19 Provider-performed microscopy (PPM) procedures.
(a) Requirement. To be categorized as a PPM procedure, the procedure must meet the criteria specified in paragraph (b) of this section.
(b) Criteria. Procedures must meet the following specifications:
(1) The examination must be personally performed by one of the following practitioners:
(i) A physician during the patient's visit on a specimen obtained from his or her own patient or from a patient of a group medical practice of which the physician is a member or an employee.
(ii) A midlevel practitioner, under the supervision of a physician or in independent practice only if authorized by the State, during the patient's visit on a specimen obtained from his or her own patient or from a patient of a clinic, group medical practice, or other health care provider of which the midlevel practitioner is a member or an employee.
(iii) A dentist during the patient's visit on a specimen obtained from his or her own patient or from a patient of a group dental practice of which the dentist is a member or an employee.
(2) The procedure must be categorized as moderately complex.
(3) The primary instrument for performing the test is the microscope, limited to bright-field or phase-contrast microscopy.
(4) The specimen is labile or delay in performing the test could compromise the accuracy of the test result.
(5) Control materials are not available to monitor the entire testing process.
(6) Limited specimen handling or processing is required.
(c) Provider-performed microscopy (PPM) examinations. A laboratory may qualify to perform tests under this section if it restricts PPM examinations to one or more of the following procedures (or additional procedures added to this list as provided under paragraph (d) of this section), waived tests and no others:
(1) All direct wet mount preparations for the presence or absence of bacteria, fungi, parasites, and human cellular elements.
(2) All potassium hydroxide (KOH) preparations.
(3) Pinworm examinations.
(4) Fern tests.
(5) Post-coital direct, qualitative examinations of vaginal or cervical mucous.
(6) Urine sediment examinations.
(7) Nasal smears for granulocytes.
(8) Fecal leukocyte examinations.
(9) Qualitative semen analysis (limited to the presence or absence of sperm and detection of motility).
(d) Revisions to criteria and the list of PPM procedures.
(1) The CLIAC conducts reviews upon HHS' request and recommends to HHS revisions to the criteria for categorization of procedures.
(2) HHS determines whether a laboratory procedure meets the criteria listed under paragraph (b) of this section for a PPM procedure. Revisions to the list of PPM procedures proposed by HHS are published in the Federal Register as a notice with an opportunity for public comment.
(e) Laboratory requirements. Laboratories eligible to perform PPM examinations must—
(1) Meet the applicable requirements in subpart C or subpart D, and subparts F, H, J, K, and M of this part.
(2) Be subject to inspection as specified under subpart Q of this part.
[60 FR 20044, Apr. 24, 1995; 68 FR 50723, Aug. 22, 2003]
§ 493.20 Laboratories performing tests of moderate complexity.
(a) A laboratory may qualify for a certificate to perform tests of moderate complexity provided that it restricts its test performance to waived tests or examinations and one or more tests or examinations meeting criteria for tests of moderate complexity including the subcategory of PPM procedures.
(b) A laboratory that performs tests or examinations of moderate complexity must meet the applicable requirements in subpart C or subpart D, and subparts F, H, J, K, M, and Q of this part. Under a registration certificate or certificate of compliance, laboratories also performing PPM procedures must meet the inspection requirements at §§ 493.1773 and 493.1777.
(c) If the laboratory also performs waived tests, compliance with subparts H, J, K, and M of this part is not applicable to the waived tests. However, the laboratory must comply with the requirements in §§ 493.15(e), 493.1773, and 493.1775.
[60 FR 20044, Apr. 24, 1995, as amended at 68 FR 3702, Jan. 24, 2003; 68 FR 50723, Aug. 22, 2003]
§ 493.25 Laboratories performing tests of high complexity.
(a) A laboratory must obtain a certificate for tests of high complexity if it performs one or more tests that meet the criteria for tests of high complexity as specified in § 493.17(a).
(b) A laboratory performing one or more tests of high complexity must meet the applicable requirements of subpart C or subpart D, and subparts F, H, J, K, M, and Q of this part.
(c) If the laboratory also performs tests of moderate complexity, the applicable requirements of subparts H, J, K, M, and Q of this part must be met. Under a registration certificate or certificate of compliance, PPM procedures must meet the inspection requirements at §§ 493.1773 and 493.1777.
(d) If the laboratory also performs waived tests, the requirements of subparts H, J, K, and M are not applicable to the waived tests. However, the laboratory must comply with the requirements in §§ 493.15(e), 493.1773, and 493.1775.
[57 FR 7139, Feb. 28, 1992, as amended at 60 FR 20044, Apr. 24, 1995; 68 FR 3702, Jan. 24, 2003; 68 FR 50723, Aug. 22, 2003]
Subpart B - Certificate of Waiver
Source:
57 FR 7142, Feb. 28, 1992, unless otherwise noted.
§ 493.35 Application for a certificate of waiver.
(a) Filing of application. Except as specified in paragraph (b) of this section, a laboratory performing only one or more waived tests listed in § 493.15 must file a separate application for each laboratory location.
(b) Exceptions.
(1) Laboratories that are not at a fixed location, that is, laboratories that move from testing site to testing site, such as mobile units providing laboratory testing, health screening fairs, or other temporary testing locations may be covered under the certificate of the designated primary site or home base, using its address.
(2) Not-for-profit or Federal, State, or local government laboratories that engage in limited (not more than a combination of 15 moderately complex or waived tests per certificate) public health testing may file a single application.
(3) Laboratories within a hospital that are located at contiguous buildings on the same campus and under common direction may file a single application or multiple applications for the laboratory sites within the same physical location or street address.
(c) Application format and contents. The application must—
(1) Be made to HHS or its designee on a form or forms prescribed by HHS;
(2) Be signed by an owner, or by an authorized representative of the laboratory who attests that the laboratory will be operated in accordance with requirements established by the Secretary under section 353 of the PHS Act; and
(3) Describe the characteristics of the laboratory operation and the examinations and other test procedures performed by the laboratory including—
(i) The name and the total number of test procedures and examinations performed annually (excluding tests the laboratory may run for quality control, quality assurance or proficiency testing purposes;
(ii) The methodologies for each laboratory test procedure or examination performed, or both; and
(iii) The qualifications (educational background, training, and experience) of the personnel directing and supervising the laboratory and performing the laboratory examinations and test procedures.
(d) Access requirements. Laboratories that perform one or more waived tests listed in § 493.15(c) and no other tests must meet the following conditions:
(1) Make records available and submit reports to HHS as HHS may reasonably require to determine compliance with this section and § 493.15(e);
(2) Agree to permit announced and unannounced inspections by HHS in accordance with subpart Q of this part under the following circumstances:
(i) When HHS has substantive reason to believe that the laboratory is being operated in a manner that constitutes an imminent and serious risk to human health.
(ii) To evaluate complaints from the public.
(iii) On a random basis to determine whether the laboratory is performing tests not listed in § 493.15.
(iv) To collect information regarding the appropriateness of waiver of tests listed in § 493.15.
(e) Denial of application. If HHS determines that the application for a certificate of waiver is to be denied, HHS will—
(1) Provide the laboratory with a written statement of the grounds on which the denial is based and an opportunity for appeal, in accordance with the procedures set forth in subpart R of this part;
(2) Notify a laboratory that has its application for a certificate of waiver denied that it cannot operate as a laboratory under the PHS Act unless the denial is overturned at the conclusion of the administrative appeals process provided by subpart R; and
(3) Notify the laboratory that it is not eligible for payment under the Medicare and Medicaid programs.
[57 FR 7142, Feb. 28, 1992, as amended at 58 FR 5222, Jan. 19, 1993; 60 FR 20044, Apr. 24, 1995]
§ 493.37 Requirements for a certificate of waiver.
(a) HHS will issue a certificate of waiver to a laboratory only if the laboratory meets the requirements of § 493.35.
(b) Laboratories issued a certificate of waiver—
(1) Are subject to the requirements of this subpart and § 493.15(e) of subpart A of this part; and
(2) Must permit announced or unannounced inspections by HHS in accordance with subpart Q of this part.
(c) Laboratories must remit the certificate of waiver fee specified in subpart F of this part.
(d) In accordance with subpart R of this part, HHS will suspend or revoke or limit a laboratory's certificate of waiver for failure to comply with the requirements of this subpart. In addition, failure to meet the requirements of this subpart will result in suspension or denial of payments under Medicare and Medicaid in accordance with subpart R of this part.
(e)
(1) A certificate of waiver issued under this subpart is valid for no more than 2 years. In the event of a non-compliance determination resulting in HHS action to revoke, suspend, or limit the laboratory's certificate of waiver, HHS will provide the laboratory with a statement of grounds on which the determination of non-compliance is based and offer an opportunity for appeal as provided in subpart R of this part.
(2) If the laboratory requests a hearing within the time specified by HHS, it retains its certificate of waiver or reissued certificate of waiver until a decision is made by an administrative law judge, as specified in subpart R of this part, except when HHS finds that conditions at the laboratory pose an imminent and serious risk to human health.
(3) For laboratories receiving payment from the Medicare or Medicaid program, such payments will be suspended on the effective date specified in the notice to the laboratory of a non-compliance determination even if there has been no appeals decision issued.
(f) A laboratory seeking to renew its certificate of waiver must—
(1) Complete the renewal application prescribed by HHS and return it to HHS not less than 9 months nor more than 1 year before the expiration of the certificate; and
(g) A laboratory with a certificate of waiver that wishes to perform examinations or tests not listed in the waiver test category must meet the requirements set forth in subpart C or subpart D of this part, as applicable.
[57 FR 7142, Feb. 28, 1992, as amended at 58 FR 5222, Jan. 19, 1993; 60 FR 20045, Apr. 24, 1995]
§ 493.39 Notification requirements for laboratories issued a certificate of waiver.
Laboratories performing one or more tests listed in § 493.15 and no others must notify HHS or its designee—
(a) Before performing and reporting results for any test or examination that is not specified under § 493.15 for which the laboratory does not have the appropriate certificate as required in subpart C or subpart D of this part, as applicable; and
(b) Within 30 days of any change(s) in—
(1) Ownership;
(2) Name;
(3) Location; or
(4) Director.
[57 FR 7142, Feb. 28, 1992, as amended at 60 FR 20045, Apr. 24, 1995]
§ 493.41 Condition: Reporting of SARS-CoV-2 test results.
During the Public Health Emergency, as defined in § 400.200 of this chapter, each laboratory that performs a test that is intended to detect SARS-CoV-2 or to diagnose a possible case of COVID-19 (hereinafter referred to as a “SARS-CoV-2 test”) must report SARS-CoV-2 test results to the Secretary in such form and manner, and at such timing and frequency, as the Secretary may prescribe.
[85 FR 54873, Sept. 2, 2020]
Subpart C - Registration Certificate, Certificate for Provider-performed Microscopy Procedures, and Certificate of Compliance
Source:
57 FR 7143, Feb. 28, 1992, unless otherwise noted.
§ 493.43 Application for registration certificate, certificate for provider-performed microscopy (PPM) procedures, and certificate of compliance.
(a) Filing of application. Except as specified in paragraph (b) of this section, all laboratories performing nonwaived testing must file a separate application for each laboratory location.
(b) Exceptions.
(1) Laboratories that are not at a fixed location, that is, laboratories that move from testing site to testing site, such as mobile units providing laboratory testing, health screening fairs, or other temporary testing locations may be covered under the certificate of the designated primary site or home base, using its address.
(2) Not-for-profit or Federal, State, or local government laboratories that engage in limited (not more than a combination of 15 moderately complex or waived tests per certificate) public health testing may file a single application.
(3) Laboratories within a hospital that are located at contiguous buildings on the same campus and under common direction may file a single application or multiple applications for the laboratory sites within the same physical location or street address.
(c) Application format and contents. The application must—
(1) Be made to HHS or its designee on a form or forms prescribed by HHS;
(2) Be signed by an owner, or by an authorized representative of the laboratory who attests that the laboratory will be operated in accordance with the requirements established by the Secretary under section 353 of the Public Health Service Act; and
(3) Describe the characteristics of the laboratory operation and the examinations and other test procedures performed by the laboratory including—
(i) The name and total number of test procedures and examinations performed annually (excluding waived tests or tests for quality control, quality assurance or proficiency testing purposes);
(ii) The methodologies for each laboratory test procedure or examination performed, or both;
(iii) The qualifications (educational background, training, and experience) of the personnel directing and supervising the laboratory and performing the examinations and test procedures.
(d) Access and reporting requirements. All laboratories must make records available and submit reports to HHS as HHS may reasonably require to determine compliance with this section.
[57 FR 7143, Feb. 28, 1992, as amended at 58 FR 5222, Jan. 19, 1993; 58 FR 39155, July 22, 1993; 60 FR 20045, Apr. 24, 1995; 68 FR 3702, Jan. 24, 2003]
§ 493.45 Requirements for a registration certificate.
Laboratories performing only waived tests, PPM procedures, or any combination of these tests, are not required to obtain a registration certificate.
(a) A registration certificate is required—
(1) Initially for all laboratories performing test procedures of moderate complexity (other than the subcategory of PPM procedures) or high complexity, or both; and
(2) For all laboratories that have been issued a certificate of waiver or certificate for PPM procedures that intend to perform tests of moderate or high complexity, or both, in addition to those tests listed in § 493.15(c) or specified as PPM procedures.
(b) HHS will issue a registration certificate if the laboratory—
(1) Complies with the requirements of § 493.43;
(2) Agrees to notify HHS or its designee within 30 days of any changes in ownership, name, location, director or technical supervisor (laboratories performing high complexity testing only);
(3) Agrees to treat proficiency testing samples in the same manner as it treats patient specimens; and
(4) Remits the fee for the registration certificate, as specified in subpart F of this part.
(c) Prior to the expiration of the registration certificate, a laboratory must—
(1) Remit the certificate fee specified in subpart F of this part;
(2) Be inspected by HHS as specified in subpart Q of this part; and
(3) Demonstrate compliance with the applicable requirements of this subpart and subparts H, J, K, M, and Q of this part.
(d) In accordance with subpart R of this part, HHS will initiate suspension or revocation of a laboratory's registration certificate and will deny the laboratory's application for a certificate of compliance for failure to comply with the requirements set forth in this subpart. HHS may also impose certain alternative sanctions. In addition, failure to meet the requirements of this subpart will result in suspension of payments under Medicare and Medicaid as specified in subpart R of this part.
(e) A registration certificate is—
(1) Valid for a period of no more than two years or until such time as an inspection to determine program compliance can be conducted, whichever is shorter; and
(2) Not renewable; however, the registration certificate may be reissued if compliance has not been determined by HHS prior to the expiration date of the registration certificate.
(f) In the event of a noncompliance determination resulting in an HHS denial of a laboratory's certificate of compliance application, HHS will provide the laboratory with a statement of grounds on which the noncompliance determination is based and offer an opportunity for appeal as provided in subpart R.
(g) If the laboratory requests a hearing within the time specified by HHS, it retains its registration certificate or reissued registration certificate until a decision is made by an administrative law judge as provided in subpart R of this part, except when HHS finds that conditions at the laboratory pose an imminent and serious risk to human health.
(h) For laboratories receiving payment from the Medicare or Medicaid program, such payments will be suspended on the effective date specified in the notice to the laboratory of denial of the certificate application even if there has been no appeals decision issued.
[57 FR 7143, Feb. 28, 1992, as amended at 58 FR 5223, Jan. 19, 1993; 60 FR 20045, Apr. 24, 1995; 68 FR 3702, Jan. 24, 2003]
§ 493.47 Requirements for a certificate for provider-performed microscopy (PPM) procedures.
(a) A certificate for PPM procedures is required—
(1) Initially for all laboratories performing test procedures specified as PPM procedures; and
(2) For all certificate of waiver laboratories that intend to perform only test procedures specified as PPM procedures in addition to those tests listed in § 493.15(c).
(b) HHS will issue a certificate for PPM procedures if the laboratory—
(1) Complies with the requirements of § 493.43; and
(2) Remits the fee for the certificate, as specified in subpart F of this part.
(c) Laboratories issued a certificate for PPM procedures are subject to—
(1) The notification requirements of § 493.53;
(2) The applicable requirements of this subpart and subparts H, J, K, and M of this part; and
(3) Inspection only under the circumstances specified under §§ 493.1773 and 493.1775, but are not routinely inspected to determine compliance with the requirements specified in paragraphs (c) (1) and (2) of this section.
(d) In accordance with subpart R of this part, HHS will initiate suspension, limitation, or revocation of a laboratory's certificate for PPM procedures for failure to comply with the applicable requirements set forth in this subpart. HHS may also impose certain alternative sanctions. In addition, failure to meet the requirements of this subpart may result in suspension of all or part of payments under Medicare and Medicaid, as specified in subpart R of this part.
(e) A certificate for PPM procedures is valid for a period of no more than 2 years.
[58 FR 5223, Jan. 19, 1993, as amended at 60 FR 20045, Apr. 24, 1995; 68 FR 3702, Jan. 24, 2003; 68 FR 50723, Aug. 22, 2003]
§ 493.49 Requirements for a certificate of compliance.
A certificate of compliance may include any combination of tests categorized as high complexity or moderate complexity or listed in § 493.15(c) as waived tests. Moderate complexity tests may include those specified as PPM procedures.
(a) HHS will issue a certificate of compliance to a laboratory only if the laboratory—
(2) Remits the certificate fee specified in subpart F of this part; and
(3) Meets the applicable requirements of this subpart and subparts H, J, K, M, and Q of this part.
(b) Laboratories issued a certificate of compliance—
(1) Are subject to the notification requirements of § 493.51; and
(2) Must permit announced or unannounced inspections by HHS in accordance with subpart Q of this part—
(i) To determine compliance with the applicable requirements of this part;
(ii) To evaluate complaints;
(iii) When HHS has substantive reason to believe that tests are being performed, or the laboratory is being operated in a manner that constitutes an imminent and serious risk to human health; and
(iv) To collect information regarding the appropriateness of tests listed in § 493.15 or tests categorized as moderate complexity (including the subcategory) or high complexity.
(c) Failure to comply with the requirements of this subpart will result in—
(1) Suspension, revocation or limitation of a laboratory's certificate of compliance in accordance with subpart R of this part; and
(2) Suspension or denial of payments under Medicare and Medicaid in accordance with subpart R of this part.
(d) A certificate of compliance issued under this subpart is valid for no more than 2 years.
(e) In the event of a noncompliance determination resulting in an HHS action to revoke, suspend or limit the laboratory's certificate of compliance, HHS will—
(1) Provide the laboratory with a statement of grounds on which the determination of noncompliance is based; and
(2) Offer an opportunity for appeal as provided in subpart R of this part. If the laboratory requests a hearing within 60 days of the notice of sanction, it retains its certificate of compliance or reissued certificate of compliance until a decision is made by an administrative law judge (ALJ) as provided in subpart R of this part, except when HHS finds that conditions at the laboratory pose an imminent and serious risk to human health or when the criteria at § 493.1840(a) (4) and (5) are met.
(f) For laboratories receiving payment from the Medicare or Medicaid program, such payments will be suspended on the effective date specified in the notice to the laboratory of a noncompliance determination even if there has been no appeals decision issued.
(g) A laboratory seeking to renew its certificate of compliance must—
(1) Complete and return the renewal application to HHS 9 to 12 months prior to the expiration of the certificate of compliance; and
(2) Meet the requirements of § 493.43 and paragraphs (a)(2) and (b)(2) of this section.
(h) If HHS determines that the application for the renewal of a certificate of compliance must be denied or limited, HHS will notify the laboratory in writing of the—
(1) Basis for denial of the application; and
(2) Opportunity for appeal as provided in subpart R of this part.
(i) If the laboratory requests a hearing within the time period specified by HHS, the laboratory retains its certificate of compliance or reissued certificate of compliance until a decision is made by an ALJ as provided in subpart R, except when HHS finds that conditions at the laboratory pose an imminent and serious risk to human health.
(j) For laboratories receiving payment from the Medicare or Medicaid program, such payments will be suspended on the effective date specified in the notice to the laboratory of nonrenewal of the certificate of compliance even if there has been no appeals decision issued.
[60 FR 20045, Apr. 24, 1995, as amended at 68 FR 3702, Jan. 24, 2003]
§ 493.51 Notification requirements for laboratories issued a certificate of compliance.
Laboratories issued a certificate of compliance must meet the following conditions:
(a) Notify HHS or its designee within 30 days of any change in—
(1) Ownership;
(2) Name;
(3) Location;
(4) Director; or
(5) Technical supervisor (laboratories performing high complexity only).
(b) Notify HHS no later than 6 months after performing any test or examination within a specialty or subspecialty area that is not included on the laboratory's certificate of compliance, so that compliance with requirements can be determined.
(c) Notify HHS no later than 6 months after any deletions or changes in test methodologies for any test or examination included in a specialty or subspecialty, or both, for which the laboratory has been issued a certificate of compliance.
[57 FR 7143, Feb. 28, 1992, as amended at 60 FR 20046, Apr. 24, 1995]
§ 493.53 Notification requirements for laboratories issued a certificate for provider-performed microscopy (PPM) procedures.
Laboratories issued a certificate for PPM procedures must notify HHS or its designee—
(a) Before performing and reporting results for any test of moderate or high complexity, or both, in addition to tests specified as PPM procedures or any test or examination that is not specified under § 493.15(c), for which it does not have a registration certificate as required in subpart C or subpart D, as applicable, of this part; and
(b) Within 30 days of any change in—
(1) Ownership;
(2) Name;
(3) Location; or
(4) Director.
[58 FR 5224, Jan. 19, 1993, as amended at 60 FR 20046, Apr. 24, 1995]
Subpart D - Certificate of Accreditation
Source:
57 FR 7144, Feb. 28, 1992, unless otherwise noted.
§ 493.55 Application for registration certificate and certificate of accreditation.
(a) Filing of application. A laboratory may be issued a certificate of accreditation in lieu of the applicable certificate specified in subpart B or subpart C of this part provided the laboratory—
(1) Meets the standards of a private non-profit accreditation program approved by HHS in accordance with subpart E; and
(2) Files a separate application for each location, except as specified in paragraph (b) of this section.
(b) Exceptions.
(1) Laboratories that are not at fixed locations, that is, laboratories that move from testing site to testing site, such as mobile units providing laboratory testing, health screening fairs, or other temporary testing locations may be covered under the certificate of the designated primary site or home base, using its address.
(2) Not-for-profit or Federal, State, or local government laboratories that engage in limited (not more than a combination of 15 moderately complex or waived tests per certificate) public health testing may file a single application.
(3) Laboratories within a hospital that are located at contiguous buildings on the same campus and under common direction may file a single application or multiple applications for the laboratory sites within the same physical location or street address.
(c) Application format and contents. The application must—
(1) Be made to HHS on a form or forms prescribed by HHS;
(2) Be signed by an owner or authorized representative of the laboratory who attests that the laboratory will be operated in accordance with the requirements established by the Secretary under section 353 of the Public Health Service Act; and
(3) Describe the characteristics of the laboratory operation and the examinations and other test procedures performed by the laboratory including—
(i) The name and total number of tests and examinations performed annually (excluding waived tests and tests for quality control, quality assurance or proficiency testing purposes);
(ii) The methodologies for each laboratory test procedure or examination performed, or both; and
(iii) The qualifications (educational background, training, and experience) of the personnel directing and supervising the laboratory and performing the laboratory examinations and test procedures.
(d) Access and reporting requirements. All laboratories must make records available and submit reports to HHS as HHS may reasonably require to determine compliance with this section.
[57 FR 7144, Feb. 28, 1992, as amended at 58 FR 5224, Jan. 19, 1993; 58 FR 39155, July 22, 1993; 60 FR 20046, Apr. 24, 1995]
§ 493.57 Requirements for a registration certificate.
A registration certificate is required for all laboratories seeking a certificate of accreditation, unless the laboratory holds a valid certificate of compliance issued by HHS.
(a) HHS will issue a registration certificate if the laboratory—
(1) Complies with the requirements of § 493.55;
(2) Agrees to notify HHS within 30 days of any changes in ownership, name, location, director, or supervisor (laboratories performing high complexity testing only);
(3) Agrees to treat proficiency testing samples in the same manner as it treats patient specimens; and
(4) Remits the fee for the registration certificate specified in subpart F of this part.
(b)
(1) The laboratory must provide HHS with proof of accreditation by an approved accreditation program—
(i) Within 11 months of issuance of the registration certificate; or
(ii) Prior to the expiration of the certificate of compliance.
(2) If such proof of accreditation is not supplied within this timeframe, the laboratory must meet, or continue to meet, the requirements of § 493.49.
(c) In accordance with subpart R of this part, HHS will initiate suspension, revocation, or limitation of a laboratory's registration certificate and will deny the laboratory's application for a certificate of accreditation for failure to comply with the requirements set forth in this subpart. In addition, failure to meet the requirements of this subpart will result in suspension or denial of payments under Medicare and Medicaid as specified in subpart R of this part.
(d) A registration certificate is valid for a period of no more than 2 years. However, it may be reissued if the laboratory is subject to subpart C of this part, as specified in § 493.57(b)(2) and compliance has not been determined by HHS before the expiration date of the registration certificate.
(e) In the event that the laboratory does not meet the requirements of this subpart, HHS will—
(1) Deny a laboratory's request for certificate of accreditation;
(2) Notify the laboratory if it must meet the requirements for a certificate as defined in subpart C of this part;
(3) Provide the laboratory with a statement of grounds on which the application denial is based;
(4) Offer an opportunity for appeal on the application denial as provided in subpart R of this part. If the laboratory requests a hearing within the time specified by HHS, the laboratory will retain its registration certificate or reissued registration certificate until a decision is made by an administrative law judge as provided in subpart R, unless HHS finds that conditions at the laboratory pose an imminent and serious risk to human health; and
(5) For those laboratories receiving payment from the Medicare or Medicaid program, such payments will be suspended on the effective date specified in the notice to the laboratory of denial of the request even if there has been no appeals decision issued.
[57 FR 7144, Feb. 28, 1992, as amended at 60 FR 20046, Apr. 24, 1995]
§ 493.61 Requirements for a certificate of accreditation.
(a) HHS will issue a certificate of accreditation to a laboratory if the laboratory—
(1) Meets the requirements of § 493.57 or, if applicable, § 493.49 of subpart C of this part; and
(2) Remits the certificate of accreditation fee specified in subpart F of this part.
(b) Laboratories issued a certificate of accreditation must—
(1) Treat proficiency testing samples in the same manner as patient samples;
(2) Meet the requirements of § 493.63;
(3) Comply with the requirements of the approved accreditation program;
(4) Permit random sample validation and complaint inspections as required in subpart Q of this part;
(5) Permit HHS to monitor the correction of any deficiencies found through the inspections specified in paragraph (b)(4) of this section;
(6) Authorize the accreditation program to release to HHS the laboratory's inspection findings whenever HHS conducts random sample or complaint inspections; and
(7) Authorize its accreditation program to submit to HHS the results of the laboratory's proficiency testing.
(c) A laboratory failing to meet the requirements of this section—
(1) Will no longer meet the requirements of this part by virtue of its accreditation in an approved accreditation program;
(2) Will be subject to full determination of compliance by HHS;
(3) May be subject to suspension, revocation or limitation of the laboratory's certificate of accreditation or certain alternative sanctions; and
(4) May be subject to suspension of payments under Medicare and Medicaid as specified in subpart R.
(d) A certificate of accreditation issued under this subpart is valid for no more than 2 years. In the event of a non-compliance determination as a result of a random sample validation or complaint inspection, a laboratory will be subject to a full review by HHS in accordance with § 488.11 of this chapter.
(e) Failure to meet the applicable requirements of part 493, will result in an action by HHS to suspend, revoke or limit the certificate of accreditation. HHS will—
(1) Provide the laboratory with a statement of grounds on which the determination of noncompliance is based;
(2) Notify the laboratory if it is eligible to apply for a certificate as defined in subpart C of this part; and
(3) Offer an opportunity for appeal as provided in subpart R of this part.
(f) If the laboratory requests a hearing within the time frame specified by HHS—
(1) It retains its certificate of accreditation or reissued certificate of accreditation until a decision is made by an administrative law judge as provided in subpart R of this part, unless HHS finds that conditions at the laboratory pose an imminent and serious risk to human health; and
(2) For those laboratories receiving payments from the Medicare or Medicaid program, such payments will be suspended on the effective date specified in the notice to the laboratory even if there has been no appeals decision issued.
(g) In the event the accreditation organization's approval is removed by HHS, the laboratory will be subject to the applicable requirements of subpart C of this part or § 493.57.
(h) A laboratory seeking to renew its certificate of accreditation must—
(1) Complete and return the renewal application to HHS 9 to 12 months prior to the expiration of the certificate of accreditation;
(2) Meet the requirements of this subpart; and
(3) Submit the certificate of accreditation fee specified in subpart F of this part.
(i) If HHS determines that the renewal application for a certificate of accreditation is to be denied or limited, HHS will notify the laboratory in writing of—
(1) The basis for denial of the application;
(2) Whether the laboratory is eligible for a certificate as defined in subpart C of this part;
(3) The opportunity for appeal on HHS's action to deny the renewal application for certificate of accreditation as provided in subpart R of this part. If the laboratory requests a hearing within the time frame specified by HHS, it retains its certificate of accreditation or reissued certificate of accreditation until a decision is made by an administrative law judge as provided in subpart R of this part, unless HHS finds that conditions at the laboratory pose an imminent and serious risk to human health; and
(4) Suspension of payments under Medicare or Medicaid for those laboratories receiving payments under the Medicare or Medicaid programs.
[57 FR 7144, Feb. 28, 1992, as amended at 58 FR 5224, Jan. 19, 1993]
§ 493.63 Notification requirements for laboratories issued a certificate of accreditation.
Laboratories issued a certificate of accreditation must:
(a) Notify HHS and the approved accreditation program within 30 days of any changes in—
(1) Ownership;
(2) Name;
(3) Location; or
(4) Director.
(b) Notify the approved accreditation program no later than 6 months after performing any test or examination within a specialty or subspecialty area that is not included in the laboratory's accreditation, so that the accreditation organization can determine compliance and a new certificate of accreditation can be issued.
(c) Notify the accreditation program no later than 6 months after of any deletions or changes in test methodologies for any test or examination included in a specialty or subspecialty, or both, for which the laboratory has been issued a certificate of accreditation.
Subpart E - Accreditation by a Private, Nonprofit Accreditation Organization or Exemption Under an Approved State Laboratory Program
Source:
63 FR 26732, May 14, 1998, unless otherwise noted.
§ 493.551 General requirements for laboratories.
(a) Applicability. CMS may deem a laboratory to meet all applicable CLIA program requirements through accreditation by a private nonprofit accreditation program (that is, grant deemed status), or may exempt from CLIA program requirements all State licensed or approved laboratories in a State that has a State licensure program established by law, if the following conditions are met:
(1) The requirements of the accreditation organization or State licensure program are equal to, or more stringent than, the CLIA condition-level requirements specified in this part, and the laboratory would meet the condition-level requirements if it were inspected against these requirements.
(2) The accreditation program or the State licensure program meets the requirements of this subpart and is approved by CMS.
(3) The laboratory authorizes the approved accreditation organization or State licensure program to release to CMS all records and information required and permits inspections as outlined in this part.
(b) Meeting CLIA requirements by accreditation. A laboratory seeking to meet CLIA requirements through accreditation by an approved accreditation organization must do the following:
(1) Obtain a certificate of accreditation as required in subpart D of this part.
(2) Pay the applicable fees as required in subpart F of this part.
(3) Meet the proficiency testing (PT) requirements in subpart H of this part.
(4) Authorize its PT organization to furnish to its accreditation organization the results of the laboratory's participation in an approved PT program for the purpose of monitoring the laboratory's PT and for making the annual PT results, along with explanatory information required to interpret the PT results, available on a reasonable basis, upon request of any person. A laboratory that refuses to authorize release of its PT results is no longer deemed to meet the condition-level requirements and is subject to a full review by CMS, in accordance with subpart Q of this part, and may be subject to the suspension or revocation of its certificate of accreditation under § 493.1840.
(5) Authorize its accreditation organization to release to CMS or a CMS agent the laboratory's PT results that constitute unsuccessful participation in an approved PT program, in accordance with the definition of “unsuccessful participation in an approved PT program,” as specified in § 493.2 of this part, when the laboratory has failed to achieve successful participation in an approved PT program.
(6) Authorize its accreditation organization to release to CMS a notification of the actions taken by the organization as a result of the unsuccessful participation in a PT program within 30 days of the initiation of the action. Based on this notification, CMS may take an adverse action against a laboratory that fails to participate successfully in an approved PT program.
(c) Withdrawal of laboratory accreditation. After an accreditation organization has withdrawn or revoked its accreditation of a laboratory, the laboratory retains its certificate of accreditation for 45 days after the laboratory receives notice of the withdrawal or revocation of the accreditation, or the effective date of any action taken by CMS, whichever is earlier.
§ 493.553 Approval process (application and reapplication) for accreditation organizations and State licensure programs.
(a) Information required. An accreditation organization that applies or reapplies to CMS for deeming authority, or a State licensure program that applies or reapplies to CMS for exemption from CLIA program requirements of licensed or approved laboratories within the State, must provide the following information:
(1) A detailed comparison of the individual accreditation, or licensure or approval requirements with the comparable condition-level requirements; that is, a crosswalk.
(2) A detailed description of the inspection process, including the following:
(i) Frequency of inspections.
(ii) Copies of inspection forms.
(iii) Instructions and guidelines.
(iv) A description of the review and decision-making process of inspections.
(v) A statement concerning whether inspections are announced or unannounced.
(vi) A description of the steps taken to monitor the correction of deficiencies.
(3) A description of the process for monitoring PT performance, including action to be taken in response to unsuccessful participation in a CMS-approved PT program.
(4) Procedures for responding to and for the investigation of complaints against its laboratories.
(5) A list of all its current laboratories and the expiration date of their accreditation or licensure, as applicable.
(6) Procedures for making PT information available (under State confidentiality and disclosure requirements, if applicable) including explanatory information required to interpret PT results, on a reasonable basis, upon request of any person.
(b) CMS action on an application or reapplication. If CMS receives an application or reapplication from an accreditation organization, or State licensure program, CMS takes the following actions:
(1) CMS determines if additional information is necessary to make a determination for approval or denial of the application and notifies the accreditation organization or State to afford it an opportunity to provide the additional information.
(2) CMS may visit the accreditation organization or State licensure program offices to review and verify the policies and procedures represented in its application and other information, including, but not limited to, review and examination of documents and interviews with staff.
(3) CMS notifies the accreditation organization or State licensure program indicating whether CMS approves or denies the request for deeming authority or exemption, respectively, and the rationale for any denial.
(c) Duration of approval. CMS approval may not exceed 6 years.
(d) Withdrawal of application. The accreditation organization or State licensure program may withdraw its application at any time before official notification, specified at § 493.553(b)(3).
§ 493.555 Federal review of laboratory requirements.
CMS's review of an accreditation organization or State licensure program includes, but is not limited to, an evaluation of the following:
(a) Whether the organization's or State's requirements for laboratories are equal to, or more stringent than, the condition-level requirements for laboratories.
(b) The organization's or State's inspection process to determine the comparability of the full inspection and complaint inspection procedures and requirements to those of CMS, including, but not limited to, inspection frequency and the ability to investigate and respond to complaints against its laboratories.
(c) The organization's or State's agreement with CMS that requires it to do the following:
(1) Notify CMS within 30 days of the action taken, of any laboratory that has—
(i) Had its accreditation or licensure suspended, withdrawn, revoked, or limited;
(ii) In any way been sanctioned; or
(iii) Had any adverse action taken against it.
(2) Notify CMS within 10 days of any deficiency identified in an accredited or CLIA-exempt laboratory if the deficiency poses an immediate jeopardy to the laboratory's patients or a hazard to the general public.
(3) Notify CMS, within 30 days, of all newly—
(i) Accredited laboratories (or laboratories whose areas of specialty/subspecialty testing have changed); or
(ii) Licensed laboratories, including the specialty/subspecialty areas of testing.
(4) Notify each accredited or licensed laboratory within 10 days of CMS's withdrawal of the organization's deeming authority or State's exemption.
(5) Provide CMS with inspection schedules, as requested, for validation purposes.
(6) Notify CMS within 10 days of any conditional level deficiency under §§ 493.41 or 493.1100(a).
[63 FR 26732, May 14, 1998, as amended at 85 FR 54873, Sept. 2, 2020]
§ 493.557 Additional submission requirements.
(a) Specific requirements for accreditation organizations. In addition to the information specified in §§ 493.553 and 493.555, as part of the approval and review process, an accreditation organization applying or reapplying for deeming authority must also provide the following:
(1) The specialty or subspecialty areas for which the organization is requesting deeming authority and its mechanism for monitoring compliance with all requirements equivalent to condition-level requirements within the scope of the specialty or subspecialty areas.
(2) A description of the organization's data management and analysis system with respect to its inspection and accreditation decisions, including the kinds of routine reports and tables generated by the systems.
(3) Detailed information concerning the inspection process, including, but not limited to the following:
(i) The size and composition of individual accreditation inspection teams.
(ii) Qualifications, education, and experience requirements that inspectors must meet.
(iii) The content and frequency of training provided to inspection personnel, including the ability of the organization to provide continuing education and training to inspectors.
(4) Procedures for removal or withdrawal of accreditation status for laboratories that fail to meet the organization's standards.
(5) A proposed agreement between CMS and the accreditation organization with respect to the notification requirements specified in § 493.555(c).
(6) Procedures for monitoring laboratories found to be out of compliance with its requirements. (These monitoring procedures must be used only when the accreditation organization identifies noncompliance. If noncompliance is identified through validation inspections, CMS or a CMS agent monitors corrections, as authorized at § 493.565(d)).
(7) A demonstration of its ability to provide CMS with electronic data and reports in compatible code, including the crosswalk specified in § 493.553(a)(1), that are necessary for effective validation and assessment of the organization's inspection process.
(8) A demonstration of its ability to provide CMS with electronic data, in compatible code, related to the adverse actions resulting from PT results constituting unsuccessful participation in PT programs as well as data related to the PT failures, within 30 days of the initiation of adverse action.
(9) A demonstration of its ability to provide CMS with electronic data, in compatible code, for all accredited laboratories, including the area of specialty or subspecialty.
(10) Information defining the adequacy of numbers of staff and other resources.
(11) Information defining the organization's ability to provide adequate funding for performing required inspections.
(12) Any facility-specific data, upon request by CMS, which includes, but is not limited to, the following:
(i) PT results that constitute unsuccessful participation in a CMS-approved PT program.
(ii) Notification of the adverse actions or corrective actions imposed by the accreditation organization as a result of unsuccessful PT participation.
(13) An agreement to provide written notification to CMS at least 30 days in advance of the effective date of any proposed change in its requirements.
(14) An agreement to disclose any laboratory's PT results upon reasonable request by any person.
(b) Specific requirements for a State licensure program. In addition to requirements in §§ 493.553 and 493.555, as part of the approval and review process, when a State licensure program applies or reapplies for exemption from the CLIA program, the State must do the following:
(1) Demonstrate to CMS that it has enforcement authority and administrative structures and resources adequate to enforce its laboratory requirements.
(2) Permit CMS or a CMS agent to inspect laboratories in the State.
(3) Require laboratories in the State to submit to inspections by CMS or a CMS agent as a condition of licensure or approval.
(4) Agree to pay the cost of the validation program administered in that State as specified in §§ 493.645(a) and 493.646(b).
(5) Take appropriate enforcement action against laboratories found by CMS not to be in compliance with requirements equivalent to CLIA requirements.
(6) Submit for Medicare and Medicaid payment purposes, a list of the specialties and subspecialties of tests performed by each laboratory.
(7) Submit a written presentation that demonstrates the agency's ability to furnish CMS with electronic data in compatible code, including the crosswalk specified in § 493.553(a)(1).
(8) Submit a statement acknowledging that the State will notify CMS through electronic transmission of the following:
(i) Any laboratory that has had its licensure or approval revoked or withdrawn or has been in any way sanctioned by the State within 30 days of taking the action.
(ii) Changes in licensure or inspection requirements.
(iii) Changes in specialties or subspecialties under which any licensed laboratory in the State performs testing.
(9) Provide information for the review of the State's enforcement procedures for laboratories found to be out of compliance with the State's requirements.
(10) Submit information that demonstrates the ability of the State to provide CMS with the following:
(i) Electronic data and reports in compatible code with the adverse or corrective actions resulting from PT results that constitute unsuccessful participation in PT programs.
(ii) Other data that CMS determines are necessary for validation and assessment of the State's inspection process requirements.
(11) Agree to provide CMS with written notification of any changes in its licensure/approval and inspection requirements.
(12) Agree to disclose any laboratory's PT results in accordance with a State's confidentiality requirements.
(13) Agree to take the appropriate enforcement action against laboratories found by CMS not to be in compliance with requirements comparable to condition-level requirements and report these enforcement actions to CMS.
(14) If approved, reapply to CMS every 2 years to renew its exempt status and to renew its agreement to pay the cost of the CMS-administered validation program in that State.
§ 493.559 Publication of approval of deeming authority or CLIA exemption.
(a) Notice of deeming authority or exemption. CMS publishes a notice in the Federal Register when it grants deeming authority to an accreditation organization or exemption to a State licensure program.
(b) Contents of notice. The notice includes the following:
(1) The name of the accreditation organization or State licensure program.
(2) For an accreditation organization:
(i) The specific specialty or subspecialty areas for which it is granted deeming authority.
(ii) A description of how the accreditation organization provides reasonable assurance to CMS that a laboratory accredited by the organization meets CLIA requirements equivalent to those in this part and would meet CLIA requirements if the laboratory had not been granted deemed status, but had been inspected against condition-level requirements.
(3) For a State licensure program, a description of how the laboratory requirements of the State are equal to, or more stringent than, those specified in this part.
(4) The basis for granting deeming authority or exemption.
(5) The term of approval, not to exceed 6 years.
§ 493.561 Denial of application or reapplication.
(a) Reconsideration of denial.
(1) If CMS denies a request for approval, an accreditation organization or State licensure program may request, within 60 days of the notification of denial, that CMS reconsider its original application or application for renewal, in accordance with part 488, subpart D.
(2) If the accreditation organization or State licensure program requests a reconsideration of CMS's determination to deny its request for approval or reapproval, it may not submit a new application until CMS issues a final reconsideration determination.
(b) Resubmittal of a request for approval—accreditation organization. An accreditation organization may resubmit a request for approval if a final reconsideration determination is not pending and the accreditation program meets the following conditions:
(1) It has revised its accreditation program to address the rationale for denial of its previous request.
(2) It demonstrates that it can provide reasonable assurance that its accredited facilities meet condition-level requirements.
(3) It resubmits the application in its entirety.
(c) Resubmittal of request for approval—State licensure program. The State licensure program may resubmit a request for approval if a final reconsideration determination is not pending and it has taken the necessary action to address the rationale for any previous denial.
§ 493.563 Validation inspections—Basis and focus.
(a) Basis for validation inspection —
(1) Laboratory with a certificate of accreditation.
(i) CMS or a CMS agent may conduct an inspection of an accredited laboratory that has been issued a certificate of accreditation on a representative sample basis or in response to a substantial allegation of noncompliance.
(ii) CMS uses the results of these inspections to validate the accreditation organization's accreditation process.
(2) Laboratory in a State with an approved State licensure program.
(i) CMS or a CMS agent may conduct an inspection of any laboratory in a State with an approved State licensure program on a representative sample basis or in response to a substantial allegation of noncompliance.
(ii) The results of these inspections are used to validate the appropriateness of the exemption of that State's licensed or approved laboratories from CLIA program requirements.
(b) Validation inspection conducted on a representative sample basis.
(1) If CMS or a CMS agent conducts a validation inspection on a representative sample basis, the inspection is comprehensive, addressing all condition-level requirements, or it may be focused on a specific condition-level requirement.
(2) The number of laboratories sampled is sufficient to allow a reasonable estimate of the performance of the accreditation organization or State.
(c) Validation inspection conducted in response to a substantial allegation of noncompliance.
(1) If CMS or a CMS agent conducts a validation inspection in response to a substantial allegation of noncompliance, the inspection focuses on any condition-level requirement that CMS determines to be related to the allegation.
(2) If CMS or a CMS agent substantiates a deficiency and determines that the laboratory is out of compliance with any condition-level requirement, CMS or a CMS agent conducts a full CLIA inspection.
(d) Inspection of operations and offices. As part of the validation review process, CMS may conduct an onsite inspection of the operations and offices to verify the following:
(1) The accreditation organization's representations and to assess the accreditation organization's compliance with its own policies and procedures.
(2) The State's representations and to assess the State's compliance with its own policies and procedures, including verification of State enforcement actions taken on the basis of validation inspections performed by CMS or a CMS agent.
(e) Onsite inspection of an accreditation organization. An onsite inspection of an accreditation organization may include, but is not limited to, the following:
(1) A review of documents.
(2) An audit of meetings concerning the accreditation process.
(3) Evaluation of accreditation inspection results and the accreditation decision-making process.
(4) Interviews with the accreditation organization's staff.
(f) Onsite inspection of a State licensure program. An onsite inspection of a State licensure program office may include, but is not limited to, the following:
(1) A review of documents.
(2) An audit of meetings concerning the licensure or approval process.
(3) Evaluation of State inspection results and the licensure or approval decision-making process.
(4) Interviews with State employees.
§ 493.565 Selection for validation inspection—laboratory responsibilities.
A laboratory selected for a validation inspection must do the following:
(a) Authorize its accreditation organization or State licensure program, as applicable, to release to CMS or a CMS agent, on a confidential basis, a copy of the laboratory's most recent full, and any subsequent partial inspection.
(b) Authorize CMS or a CMS agent to conduct a validation inspection.
(c) Provide CMS or a CMS agent with access to all facilities, equipment, materials, records, and information that CMS or a CMS agent determines have a bearing on whether the laboratory is being operated in accordance with the requirements of this part, and permit CMS or a CMS agent to copy material or require the laboratory to submit material.
(d) If the laboratory possesses a valid certificate of accreditation, authorize CMS or a CMS agent to monitor the correction of any deficiencies found through the validation inspection.
§ 493.567 Refusal to cooperate with validation inspection.
(a) Laboratory with a certificate of accreditation.
(1) A laboratory with a certificate of accreditation that refuses to cooperate with a validation inspection by failing to comply with the requirements in § 493.565—
(i) Is subject to full review by CMS or a CMS agent, in accordance with this part; and
(ii) May be subject to suspension, revocation, or limitation of its certificate of accreditation under this part.
(2) A laboratory with a certificate of accreditation is again deemed to meet the condition-level requirements by virtue of its accreditation when the following conditions exist:
(i) The laboratory withdraws any prior refusal to authorize its accreditation organization to release a copy of the laboratory's current accreditation inspection, PT results, or notification of any adverse actions resulting from PT failure.
(ii) The laboratory withdraws any prior refusal to allow a validation inspection.
(iii) CMS finds that the laboratory meets all the condition-level requirements.
(b) CLIA-exempt laboratory. If a CLIA-exempt laboratory fails to comply with the requirements specified in § 493.565, CMS notifies the State of the laboratory's failure to meet the requirements.
§ 493.569 Consequences of a finding of noncompliance as a result of a validation inspection.
(a) Laboratory with a certificate of accreditation. If a validation inspection results in a finding that the accredited laboratory is out of compliance with one or more condition-level requirements, the laboratory is subject to—
(1) The same requirements and survey and enforcement processes applied to laboratories that are not accredited and that are found out of compliance following an inspection under this part; and
(2) Full review by CMS, in accordance with this part; that is, the laboratory is subject to the principal and alternative sanctions in § 493.1806.
(b) CLIA-exempt laboratory. If a validation inspection results in a finding that a CLIA-exempt laboratory is out of compliance with one or more condition-level requirements, CMS directs the State to take appropriate enforcement action.
§ 493.571 Disclosure of accreditation, State and CMS validation inspection results.
(a) Accreditation organization inspection results. CMS may disclose accreditation organization inspection results to the public only if the results are related to an enforcement action taken by the Secretary.
(b) State inspection results. Disclosure of State inspection results is the responsibility of the approved State licensure program, in accordance with State law.
(c) CMS validation inspection results. CMS may disclose the results of all validation inspections conducted by CMS or its agent.
§ 493.573 Continuing Federal oversight of private nonprofit accreditation organizations and approved State licensure programs.
(a) Comparability review. In addition to the initial review for determining equivalency of specified organization or State requirements to the comparable condition-level requirements, CMS reviews the equivalency of requirements in the following cases:
(1) When CMS promulgates new condition-level requirements.
(2) When CMS identifies an accreditation organization or a State licensure program whose requirements are no longer equal to, or more stringent than, condition-level requirements.
(3) When an accreditation organization or State licensure program adopts new requirements.
(4) When an accreditation organization or State licensure program adopts changes to its inspection process, as required by § 493.575(b)(1), as applicable.
(5) Every 6 years, or sooner if CMS determines an earlier review is required.
(b) Validation review. Following the end of a validation review period, CMS evaluates the validation inspection results for each approved accreditation organization and State licensure program.
(c) Reapplication procedures.
(1) Every 6 years, or sooner, as determined by CMS, an approved accreditation organization must reapply for continued approval of deeming authority and a State licensure program must reapply for continued approval of a CLIA exemption. CMS provides notice of the materials that must be submitted as part of the reapplication procedure.
(2) An accreditation organization or State licensure program that does not meet the requirements of this subpart, as determined through a comparability or validation review, must furnish CMS, upon request, with the reapplication materials CMS requests. CMS establishes a deadline by which the materials must be submitted.
(d) Notice.
(1) CMS provides written notice, as appropriate, to the following:
(i) An accreditation organization indicating that its approval may be in jeopardy if a comparability or validation review reveals that it is not meeting the requirements of this subpart and CMS is initiating a review of the accreditation organization's deeming authority.
(ii) A State licensure program indicating that its CLIA exemption may be in jeopardy if a comparability or validation review reveals that it is not meeting the requirements of this subpart and that a review is being initiated of the CLIA exemption of the State's laboratories.
(2) The notice contains the following information:
(i) A statement of the discrepancies that were found as well as other related documentation.
(ii) An explanation of CMS's review process on which the final determination is based and a description of the possible actions, as specified in § 493.575, that CMS may impose based on the findings from the comparability or validation review.
(iii) A description of the procedures available if the accreditation organization or State licensure program, as applicable, desires an opportunity to explain or justify the findings made during the comparability or validation review.
(iv) The reapplication materials that the accreditation organization or State licensure program must submit and the deadline for that submission.
§ 493.575 Removal of deeming authority or CLIA exemption and final determination review.
(a) CMS review. CMS conducts a review of the following:
(1) A deeming authority review of an accreditation organization's program if the comparability or validation review produces findings, as described at § 493.573. CMS reviews, as appropriate, the criteria described in §§ 493.555 and 493.557(a) to reevaluate whether the accreditation organization continues to meet all these criteria.
(2) An exemption review of a State's licensure program if the comparability or validation review produces findings, as described at § 493.573. CMS reviews, as appropriate, the criteria described in §§ 493.555 and 493.557(b) to reevaluate whether the licensure program continues to meet all these criteria.
(3) A review of an accreditation organization or State licensure program, at CMS's discretion, if validation review findings, irrespective of the rate of disparity, indicate widespread or systematic problems in the organization's accreditation or State's licensure process that provide evidence that the requirements, taken as a whole, are no longer equivalent to CLIA requirements, taken as a whole.
(4) A review of the accreditation organization or State licensure program whenever validation inspection results indicate a rate of disparity of 20 percent or more between the findings of the organization or State and those of CMS or a CMS agent for the following periods:
(i) One year for accreditation organizations.
(ii) Two years for State licensure programs.
(b) CMS action after review. Following the review, CMS may take the following action:
(1) If CMS determines that the accreditation organization or State has failed to adopt requirements equal to, or more stringent than, CLIA requirements, CMS may give a conditional approval for a probationary period of its deeming authority to an organization 30 days following the date of CMS's determination, or exempt status to a State within 30 days of CMS's determination, both not to exceed 1 year, to afford the organization or State an opportunity to adopt equal or more stringent requirements.
(2) If CMS determines that there are widespread or systematic problems in the organization's or State's inspection process, CMS may give conditional approval during a probationary period, not to exceed 1 year, effective 30 days following the date of the determination.
(c) Final determination. CMS makes a final determination as to whether the organization or State continues to meet the criteria described in this subpart and issues a notice that includes the reasons for the determination to the organization or State within 60 days after the end of any probationary period. This determination is based on an evaluation of any of the following:
(1) The most recent validation inspection and review findings. To continue to be approved, the organization or State must meet the criteria of this subpart.
(2) Facility-specific data, as well as other related information.
(3) The organization's or State's inspection procedures, surveyors' qualifications, ongoing education, training, and composition of inspection teams.
(4) The organization's accreditation requirements, or the State's licensure or approval requirements.
(d) Date of withdrawal of approval. CMS may withdraw its approval of the accreditation organization or State licensure program, effective 30 days from the date of written notice to the organization or State of this proposed action, if improvements acceptable to CMS have not been made during the probationary period.
(e) Continuation of validation inspections. The existence of any validation review, probationary status, or any other action, such as a deeming authority review, by CMS does not affect or limit the conduct of any validation inspection.
(f) Federal Register notice. CMS publishes a notice in the Federal Register containing a justification for removing the deeming authority from an accreditation organization, or the CLIA-exempt status of a State licensure program.
(g) Withdrawal of approval-effect on laboratory status —
(1) Accredited laboratory. After CMS withdraws approval of an accreditation organization's deeming authority, the certificate of accreditation of each affected laboratory continues in effect for 60 days after it receives notification of the withdrawal of approval.
(2) CLIA-exempt laboratory. After CMS withdraws approval of a State licensure program, the exempt status of each licensed or approved laboratory in the State continues in effect for 60 days after a laboratory receives notification from the State of the withdrawal of CMS's approval of the program.
(3) Extension. After CMS withdraws approval of an accreditation organization or State licensure program, CMS may extend the period for an additional 60 days for a laboratory if it determines that the laboratory submitted an application for accreditation to an approved accreditation organization or an application for the appropriate certificate to CMS or a CMS agent before the initial 60-day period ends.
(h) Immediate jeopardy to patients.
(1) If at any time CMS determines that the continued approval of deeming authority of any accreditation organization poses immediate jeopardy to the patients of the laboratories accredited by the organization, or continued approval otherwise constitutes a significant hazard to the public health, CMS may immediately withdraw the approval of deeming authority for that accreditation organization.
(2) If at any time CMS determines that the continued approval of a State licensure program poses immediate jeopardy to the patients of the laboratories in that State, or continued approval otherwise constitutes a significant hazard to the public health, CMS may immediately withdraw the approval of that State licensure program.
(i) Failure to pay fees. CMS withdraws the approval of a State licensure program if the State fails to pay the applicable fees, as specified in §§ 493.645(a) and 493.646(b).
(j) State refusal to take enforcement action.
(1) CMS may withdraw approval of a State licensure program if the State refuses to take enforcement action against a laboratory in that State when CMS determines it to be necessary.
(2) A laboratory that is in a State in which CMS has withdrawn program approval is subject to the same requirements and survey and enforcement processes that are applied to a laboratory that is not exempt from CLIA requirements.
(k) Request for reconsideration. Any accreditation organization or State that is dissatisfied with a determination to withdraw approval of its deeming authority or remove approval of its State licensure program, as applicable, may request that CMS reconsider the determination, in accordance with subpart D of part 488.
Subpart F - General Administration
Source:
57 FR 7138, 7213, Feb. 28, 1992, unless otherwise noted.
§ 493.602 Scope of subpart.
This subpart sets forth the methodology for determining the amount of the fees for issuing the appropriate certificate, and for determining compliance with the applicable standards of the Public Health Service Act (the PHS Act) and the Federal validation of accredited laboratories and of CLIA-exempt laboratories.
[60 FR 20047, Apr. 24, 1995]
§ 493.606 Applicability of subpart.
The rules of this subpart are applicable to those laboratories specified in § 493.3.
[58 FR 5212, Jan. 19, 1993]
§ 493.638 Certificate fees.
(a) Basic rule. Laboratories must pay a fee for the issuance of a registration certificate, certificate for PPM procedures, certificate of waiver, certificate of accreditation, or a certificate of compliance, as applicable. Laboratories must also pay a fee to reapply for a certificate for PPM procedures, certificate of waiver, certificate of accreditation, or a certificate of compliance. The total of fees collected by HHS under the laboratory program must be sufficient to cover the general costs of administering the laboratory certification program under section 353 of the PHS Act.
(1) For registration certificates and certificates of compliance, the costs include issuing the certificates, collecting the fees, evaluating and monitoring proficiency testing programs, evaluating which procedures, tests or examinations meet the criteria for inclusion in the appropriate complexity category, and implementing section 353 of the PHS Act.
(2) For a certificate of waiver, the costs include issuing the certificate, collecting the fees, determining if a certificate of waiver should be issued, evaluating which tests qualify for inclusion in the waived category, and other direct administrative costs.
(3) For a certificate for PPM procedures, the costs include issuing the certificate, collecting the fees, determining if a certificate for PPM procedures should be issued, evaluating which procedures meet the criteria for inclusion in the subcategory of PPM procedures, and other direct administrative costs.
(4) For a certificate of accreditation, the costs include issuing the certificate, collecting the fees, evaluating the programs of accrediting bodies, and other direct administrative costs.
(b) Fee amount. The fee amount is set annually by HHS on a calendar year basis and is based on the category of test complexity, or on the category of test complexity and schedules or ranges of annual laboratory test volume (excluding waived tests and tests performed for quality control, quality assurance, and proficiency testing purposes) and specialties tested, with the amounts of the fees in each schedule being a function of the costs for all aspects of general administration of CLIA as set forth in § 493.649 (b) and
(c) . This fee is assessed and payable at least biennially. The methodology used to determine the amount of the fee is found in § 493.649. The amount of the fee applicable to the issuance of the registration certificate or the issuance or renewal of the certificate for PPM procedures, certificate of waiver, certificate of accreditation, or certificate of compliance is the amount in effect at the time the application is received. Upon receipt of an application for a certificate, HHS or its designee notifies the laboratory of the amount of the required fee for the requested certificate.
[60 FR 20047, Apr. 24, 1995]
§ 493.639 Fee for revised certificate.
(a) If, after a laboratory is issued a registration certificate, it changes its name or location, the laboratory must pay a fee to cover the cost of issuing a revised registration certificate. The fee for the revised registration certificate is based on the cost to issue the revised certificate to the laboratory.
(b) A laboratory must pay a fee to cover the cost of issuing a revised certificate in any of the following circumstances:
(1) The fee for issuing an appropriate revised certificate is based on the cost to issue the revised certificate to the laboratory as follows:
(i) If a laboratory with a certificate of waiver wishes to perform tests in addition to those listed in § 493.15(c) as waived tests, it must, as set forth in § 493.638, pay an additional fee for the appropriate certificate to cover the additional testing.
(ii) If a laboratory with a certificate for PPM procedures wishes to perform tests in addition to those specified as PPM procedures or listed in § 493.15(c) as waived tests, it must, as set forth in § 493.638, pay an additional fee for the appropriate certificate to cover the additional testing.
(2) A laboratory must pay a fee to cover the cost of issuing a revised certificate when—
(i) A laboratory changes its name, location, or its director; or
(ii) A laboratory deletes services or wishes to add services and requests that its certificate be changed. (An additional fee is also required under § 493.643(d) if it is necessary to determine compliance with additional requirements.)
[57 FR 7213, Feb. 28, 1992, as amended at 60 FR 20047, Apr. 24, 1995]
§ 493.643 Fee for determination of program compliance.
(a) Fee requirement. In addition to the fee required under § 493.638, a laboratory subject to routine inspections must pay a fee to cover the cost of determining program compliance. Laboratories issued a certificate for PPM procedures, certificate of waiver, or a certificate of accreditation are not subject to this fee for routine inspections.
(b) Costs included in the fee. Included in the fee for determining program compliance is the cost of evaluating qualifications of personnel; monitoring proficiency testing; conducting onsite inspections; documenting deficiencies; evaluating laboratories' plans to correct deficiencies; and necessary administrative costs. HHS sets the fee amounts annually on a calendar year basis. Laboratories are inspected biennially; therefore, fees are assessed and payable biennially. If additional expenses are incurred to conduct follow up visits to verify correction of deficiencies, to impose sanctions, and/or for surveyor preparation for and attendance at ALJ hearings, HHS assesses an additional fee to include these costs. The additional fee is based on the actual resources and time necessary to perform the activities.
(c) Classification of laboratories that require inspection for purpose of determining amount of fee.
(1) There are ten classifications (schedules) of laboratories for the purpose of determining the fee amount a laboratory is assessed. Each laboratory is placed into one of the ten following schedules based on the laboratory's scope and volume of testing (excluding tests performed for quality control, quality assurance, and proficiency testing purposes).
(i)
(A) Schedule A Low Volume. The laboratory performs not more than 2,000 laboratory tests annually.
(B) Schedule A. The laboratory performs tests in no more than 3 specialties of service with a total annual volume of more than 2,000 but not more than 10,000 laboratory tests.
(ii) Schedule B. The laboratory performs tests in at least 4 specialties of service with a total annual volume of not more than 10,000 laboratory tests.
(iii) Schedule C. The laboratory performs tests in no more 3 specialties of service with a total annual volume of more than 10,000 but not more than 25,000 laboratory tests.
(iv) Schedule D. The laboratory performs tests in at least 4 specialties with a total annual volume of more than 10,000 but not more than 25,000 laboratory tests.
(v) Schedule E. The laboratory performs more than 25,000 but not more than 50,000 laboratory tests annually.
(vi) Schedule F. The laboratory performs more than 50,000 but not more than 75,000 laboratory tests annually.
(vii) Schedule G. The laboratory performs more than 75,000 but not more than 100,000 laboratory tests annually.
(viii) Schedule H. The laboratory performs more than 100,000 but not more than 500,000 laboratory tests annually.
(ix) Schedule I. The laboratory performs more than 500,000 but not more than 1,000,000 laboratory tests annually.
(x) Schedule J. The laboratory performs more than 1,000,000 laboratory tests annually.
(2) For purposes of determining a laboratory's classification under this section, a test is a procedure or examination for a single analyte. (Tests performed for quality control, quality assurance, and proficiency testing are excluded from the laboratory's total annual volume). Each profile (that is, group of tests) is counted as the number of separate procedures or examinations; for example, a chemistry profile consisting of 18 tests is counted as 18 separate procedures or tests.
(3) For purposes of determining a laboratory's classification under this section, the specialties and subspecialties of service for inclusion are:
(i) The specialty of Microbiology, which includes one or more of the following subspecialties:
(A) Bacteriology.
(B) Mycobacteriology.
(C) Mycology.
(D) Parasitology.
(E) Virology.
(ii) The specialty of Serology, which includes one or more of the following subspecialties:
(A) Syphilis Serology.
(B) General immunology
(iii) The specialty of Chemistry, which includes one or more of the following subspecialties:
(A) Routine chemistry.
(B) Endocrinology.
(C) Toxicology.
(D) Urinalysis.
(iv) The specialty of Hematology.
(v) The specialty of Immunohematology, which includes one or more of the following subspecialties:
(A) ABO grouping and Rh typing.
(B) Unexpected antibody detection.
(C) Compatibility testing.
(D) Unexpected antibody identification.
(vi) The specialty of Pathology, which includes the following subspecialties:
(A) Cytology.
(B) Histopathology.
(C) Oral pathology.
(vii) The specialty of Radiobioassay.
(viii) The specialty of Histocompatibility.
(ix) The specialty of Clinical Cytogenetics.
(d) Additional fees.
(1) If after a certificate of compliance is issued, a laboratory adds services and requests that its certificate be upgraded, the laboratory must pay an additional fee if, in order to determine compliance with additional requirements, it is necessary to conduct an inspection, evaluate personnel, or monitor proficiency testing performance. The additional fee is based on the actual resources and time necessary to perform the activities. HHS revokes the laboratory's certificate for failure to pay the compliance determination fee.
(2) If it is necessary to conduct a complaint investigation, impose sanctions, or conduct a hearing, HHS assesses the laboratory holding a certificate of compliance a fee to cover the cost of these activities. If a complaint investigation results in a complaint being unsubstantiated, or if an HHS adverse action is overturned at the conclusion of the administrative appeals process, the government's costs of these activities are not imposed upon the laboratory. Costs for these activities are based on the actual resources and time necessary to perform the activities and are not assessed until after the laboratory concedes the existence of deficiencies or an ALJ rules in favor of HHS. HHS revokes the laboratory's certificate of compliance for failure to pay the assessed costs.
[57 FR 7138, 7213, Feb. 28, 1992, as amended at 60 FR 20047, Apr. 24, 1995; 68 FR 3702, Jan. 24, 2003]
§ 493.645 Additional fee(s) applicable to approved State laboratory programs and laboratories issued a certificate of accreditation, certificate of waiver, or certificate for PPM procedures.
(a) Approved State laboratory programs. State laboratory programs approved by HHS are assessed a fee for the following:
(1) Costs of Federal inspections of laboratories in that State (that is, CLIA-exempt laboratories) to verify that standards are being enforced in an appropriate manner.
(2) Costs incurred for investigations of complaints against the State's CLIA-exempt laboratories if the complaint is substantiated.
(3) Costs of the State's prorata share of general overhead to develop and implement CLIA.
(b) Accredited laboratories.
(1) In addition to the certificate fee, a laboratory that is issued a certificate of accreditation is also assessed a fee to cover the cost of evaluating individual laboratories to determine overall whether an accreditation organization's standards and inspection policies are equivalent to the Federal program. All accredited laboratories share in the cost of these inspections. These costs are the same as those that are incurred when inspecting nonaccredited laboratories.
(2) If a laboratory issued a certificate of accreditation has been inspected and followup visits are necessary because of identified deficiencies, HHS assesses the laboratory a fee to cover the cost of these visits. The fee is based on the actual resources and time necessary to perform the followup visits. HHS revokes the laboratory's certificate of accreditation for failure to pay the assessed fee.
(c) If, in the case of a laboratory that has been issued a certificate of accreditation, certificate of waiver, or certificate for PPM procedures, it is necessary to conduct a complaint investigation, impose sanctions, or conduct a hearing, HHS assesses that laboratory a fee to cover the cost of these activities. Costs are based on the actual resources and time necessary to perform the activities and are not assessed until after the laboratory concedes the existence of deficiencies or an ALJ rules in favor of HHS. HHS revokes the laboratory's certificate for failure to pay the assessed costs. If a complaint investigation results in a complaint being unsubstantiated, or if an HHS adverse action is overturned at the conclusion of the administrative appeals process, the costs of these activities are not imposed upon the laboratory.
[60 FR 20047, Apr. 24, 1995]
§ 493.646 Payment of fees.
(a) Except for CLIA-exempt laboratories, all laboratories are notified in writing by HHS or its designee of the appropriate fee(s) and instructions for submitting the fee(s), including the due date for payment and where to make payment. The appropriate certificate is not issued until the applicable fees have been paid.
(b) For State-exempt laboratories, HHS estimates the cost of conducting validation surveys within the State for a 2-year period. HHS or its designee notifies the State by mail of the appropriate fees, including the due date for payment and the address of the United States Department of Treasury designated commercial bank to which payment must be made. In addition, if complaint investigations are conducted in laboratories within these States and are substantiated, HHS bills the State(s) the costs of the complaint investigations.
[57 FR 7138, 7213, Feb. 28, 1992, as amended at 60 FR 20048, Apr. 24, 1995]
§ 493.649 Methodology for determining fee amount.
(a) General rule. The amount of the fee in each schedule for compliance determination inspections is based on the average hourly rate (which includes the costs to perform the required activities and necessary administration costs) multiplied by the average number of hours required or, if activities are performed by more than one of the entities listed in paragraph (b) of this section, the sum of the products of the applicable hourly rates multiplied by the average number of hours required by the entity to perform the activity. The fee for issuance of the registration certificate or certificate of compliance is based on the laboratory's scope and volume of testing.
(b) Determining average hourly rates used in fee schedules. Three different entities perform activities related to the issuance or reissuance of any certificate. HHS determines the average hourly rates for the activities of each of these entities.
(1) State survey agencies. The following costs are included in determining an average hourly rate for the activities performed by State survey agencies:
(i) The costs incurred by the State survey agencies in evaluating personnel qualifications and monitoring each laboratory's participation in an approved proficiency testing program. The cost of onsite inspections and monitoring activities is the hourly rate derived as a result of an annual budget negotiation process with each State. The hourly rate encompasses salary costs (as determined by each State's civil service pay scale) and fringe benefit costs to support the required number of State inspectors, management and direct support staff.
(ii) Travel costs necessary to comply with each State's administrative requirements and other direct costs such as equipment, printing, and supplies. These costs are established based on historical State requirements.
(iii) Indirect costs as negotiated by HHS.
(2) Federal agencies. The hourly rate for activities performed by Federal agencies is the most recent average hourly cost to HHS to staff and support a full time equivalent employee. Included in this cost are salary and fringe benefit costs, necessary administrative costs, such as printing, training, postage, express mail, supplies, equipment, computer system and building service charges associated with support services provided by organizational components such as a computer center, and any other oversight activities necessary to support the program.
(3) HHS contractors. The hourly rate for activities performed by HHS contractors is the average hourly rate established for contractor assistance based on an independent government cost estimate for the required workload. This rate includes the cost of contractor support to provide proficiency testing programs to laboratories that do not participate in an approved proficiency testing program, provide specialized assistance in the evaluation of laboratory performance in an approved proficiency testing program, perform assessments of cytology testing laboratories, conduct special studies, bill and collect fees, issue certificates, establish accounting, monitoring and reporting systems, and assist with necessary surveyor training.
(c) Determining number of hours. The average number of hours used to determine the overall fee in each schedule is HHS's estimate, based on historical experience, of the average time needed by each entity to perform the activities for which it is responsible.
[57 FR 7138, 7213, Feb. 28, 1992, as amended at 60 FR 20048, Apr. 24, 1995]
Subpart G [Reserved]
Subpart H - Participation in Proficiency Testing for Laboratories Performing Nonwaived Testing
Source:
57 FR 7146, Feb. 28, 1992, unless otherwise noted.
§ 493.801 Condition: Enrollment and testing of samples.
Each laboratory must enroll in a proficiency testing (PT) program that meets the criteria in subpart I of this part and is approved by HHS. The laboratory must enroll in an approved program or programs for each of the specialties and subspecialties for which it seeks certification. The laboratory must test the samples in the same manner as patients' specimens. For laboratories subject to 42 CFR part 493 published on March 14, 1990 (55 FR 9538) prior to September 1, 1992, the rules of this subpart are effective on September 1, 1992. For all other laboratories, the rules of this subpart are effective January 1, 1994.
(a) Standard; Enrollment. The laboratory must -
(1) Notify HHS of the approved program or programs in which it chooses to participate to meet proficiency testing requirements of this subpart.
(2)
(i) Designate the program(s) to be used for each specialty, subspecialty, and analyte or test to determine compliance with this subpart if the laboratory participates in more than one proficiency testing program approved by CMS; and
(ii) For those tests performed by the laboratory that are not included in subpart I of this part, a laboratory must establish and maintain the accuracy of its testing procedures, in accordance with § 493.1236(c)(1).
(3) For each specialty, subspecialty and analyte or test, participate in one approved proficiency testing program or programs, for one year before designating a different program and must notify CMS before any change in designation; and
(4) Authorize the proficiency testing program to release to HHS all data required to -
(i) Determine the laboratory's compliance with this subpart; and
(ii) Make PT results available to the public as required in section 353(f)(3)(F) of the Public Health Service Act.
(b) Standard: Testing of proficiency testing samples. The laboratory must examine or test, as applicable, the proficiency testing samples it receives from the proficiency testing program in the same manner as it tests patient specimens. This testing must be conducted in conformance with paragraph (b)(4) of this section. If the laboratory's patient specimen testing procedures would normally require reflex, distributive, or confirmatory testing at another laboratory, the laboratory should test the proficiency testing sample as it would a patient specimen up until the point it would refer a patient specimen to a second laboratory for any form of further testing.
(1) The samples must be examined or tested with the laboratory's regular patient workload by personnel who routinely perform the testing in the laboratory, using the laboratory's routine methods. The individual testing or examining the samples and the laboratory director must attest to the routine integration of the samples into the patient workload using the laboratory's routine methods.
(2) The laboratory must test samples the same number of times that it routinely tests patient samples.
(3) Laboratories that perform tests on proficiency testing samples must not engage in any inter-laboratory communications pertaining to the results of proficiency testing sample(s) until after the date by which the laboratory must report proficiency testing results to the program for the testing event in which the samples were sent. Laboratories with multiple testing sites or separate locations must not participate in any communications or discussions across sites/locations concerning proficiency testing sample results until after the date by which the laboratory must report proficiency testing results to the program.
(4) The laboratory must not send proficiency testing samples or portions of proficiency testing samples to another laboratory for any analysis for which it is certified to perform in its own laboratory. Any laboratory that CMS determines intentionally referred a proficiency testing sample to another laboratory for analysis may have its certification revoked for at least 1 year. If CMS determines that a proficiency testing sample was referred to another laboratory for analysis, but the requested testing was limited to reflex, distributive, or confirmatory testing that, if the sample were a patient specimen, would have been in full conformance with written, legally accurate and adequate standard operating procedures for the laboratory's testing of patient specimens, and if the proficiency testing referral is not a repeat proficiency testing referral, CMS will consider the referral to be improper and subject to alternative sanctions in accordance with § 493.1804(c), but not intentional. Any laboratory that receives a proficiency testing sample from another laboratory for testing must notify CMS of the receipt of that sample regardless of whether the referral was made for reflex or confirmatory testing, or any other reason.
(5) The laboratory must document the handling, preparation, processing, examination, and each step in the testing and reporting of results for all proficiency testing samples. The laboratory must maintain a copy of all records, including a copy of the proficiency testing program report forms used by the laboratory to record proficiency testing results including the attestation statement provided by the PT program, signed by the analyst and the laboratory director, documenting that proficiency testing samples were tested in the same manner as patient specimens, for a minimum of two years from the date of the proficiency testing event.
(6) PT is required for only the test system, assay, or examination used as the primary method for patient testing during the PT event.
[57 FR 7146, Feb. 28, 1992, as amended at 58 FR 5228, Jan. 19, 1993; 68 FR 3702, Jan. 24, 2003; 79 FR 27157, May 12, 2014]
§ 493.803 Condition: Successful participation.
(a) Each laboratory performing nonwaived testing must successfully participate in a proficiency testing program approved by CMS, if applicable, as described in subpart I of this part for each specialty, subspecialty, and analyte or test in which the laboratory is certified under CLIA.
(b) Except as specified in paragraph (c) of this section, if a laboratory fails to participate successfully in proficiency testing for a given specialty, subspecialty, analyte or test, as defined in this section, or fails to take remedial action when an individual fails gynecologic cytology, CMS imposes sanctions, as specified in subpart R of this part.
(c) If a laboratory fails to perform successfully in a CMS-approved proficiency testing program, for the initial unsuccessful performance, CMS may direct the laboratory to undertake training of its personnel or to obtain technical assistance, or both, rather than imposing alternative or principle sanctions except when one or more of the following conditions exists:
(1) There is immediate jeopardy to patient health and safety.
(2) The laboratory fails to provide CMS or a CMS agent with satisfactory evidence that it has taken steps to correct the problem identified by the unsuccessful proficiency testing performance.
(3) The laboratory has a poor compliance history.
[57 FR 7146, Feb. 28, 1992, as amended at 60 FR 20048, Apr. 24, 1995; 63 FR 26737, May 14, 1998; 68 FR 3702, Jan. 24, 2003]
§ 493.807 Condition: Reinstatement of laboratories performing nonwaived testing.
(a) If a laboratory's certificate is suspended or limited or its Medicare or Medicaid approval is cancelled or its Medicare or Medicaid payments are suspended because it fails to participate successfully in proficiency testing for one or more specialties, subspecialties, analyte or test, or voluntarily withdraws its certification under CLIA for the failed specialty, subspecialty, or analyte, the laboratory must then demonstrate sustained satisfactory performance on two consecutive proficiency testing events, one of which may be on site, before CMS will consider it for reinstatement for certification and Medicare or Medicaid approval in that specialty, subspecialty, analyte or test.
(b) The cancellation period for Medicare and Medicaid approval or period for suspension of Medicare or Medicaid payments or suspension or limitation of certification under CLIA for the failed specialty, subspecialty, or analyte or test is for a period of not less than six months from the date of cancellation, limitation or suspension of the CLIA certificate.
[58 FR 5228, Jan. 19, 1993, as amended at 60 FR 20048, Apr. 24, 1995]
Proficiency Testing by Specialty and Subspecialty for Laboratories Performing Tests of Moderate Complexity (Including the Subcategory), High Complexity, or Any Combination of These Tests
§ 493.821 Condition: Microbiology.
The specialty of microbiology includes, for purposes of proficiency testing, the subspecialties of bacteriology, mycobacteriology, mycology, parasitology and virology.
§ 493.823 Standard; Bacteriology.
(a) Failure to attain an overall testing event score of at least 80 percent is unsatisfactory performance.
(b) Failure to participate in a testing event is unsatisfactory performance and results in a score of 0 for the testing event. Consideration may be given to those laboratories failing to participate in a testing event only if—
(1) Patient testing was suspended during the time frame allotted for testing and reporting proficiency testing results;
(2) The laboratory notifies the inspecting agency and the proficiency testing program within the time frame for submitting proficiency testing results of the suspension of patient testing and the circumstances associated with failure to perform tests on proficiency testing samples; and
(3) The laboratory participated in the previous two proficiency testing events.
(c) Failure to return proficiency testing results to the proficiency testing program within the time frame specified by the program is unsatisfactory performance and results in a score of 0 for the testing event.
(d)
(1) For any unsatisfactory testing event for reasons other than a failure to participate, the laboratory must undertake appropriate training and employ the technical assistance necessary to correct problems associated with a proficiency testing failure.
(2) Remedial action must be taken and documented, and the documentation must be maintained by the laboratory for two years from the date of participation in the proficiency testing event.
(e) Failure to achieve an overall testing event score of satisfactory performance for two consecutive testing events or two out of three consecutive testing events is unsuccessful performance.
§ 493.825 Standard; Mycobacteriology.
(a) Failure to attain an overall testing event score of at least 80 percent is unsatisfactory performance.
(b) Failure to participate in a testing event is unsatisfactory performance and results in a score of 0 for the testing event. Consideration may be given to those laboratories failing to participate in a testing event only if—
(1) Patient testing was suspended during the time frame allotted for testing and reporting proficiency testing results;
(2) The laboratory notifies the inspecting agency and the proficiency testing program within the time frame for submitting proficiency testing results of the suspension of patient testing and the circumstances associated with failure to perform tests on proficiency testing samples; and
(3) The laboratory participated in the previous two proficiency testing events.
(c) Failure to return proficiency testing results to the proficiency testing program within the time frame specified by the program is unsatisfactory performance and results in a score of 0 for the testing event.
(d)
(1) For any unsatisfactory testing event for reasons other than a failure to participate, the laboratory must undertake appropriate training and employ the technical assistance necessary to correct problems associated with a proficiency testing failure.
(2) Remedial action must be taken and documented, and the documentation must be maintained by the laboratory for two years from the date of participation in the proficiency testing event.
(e) Failure to achieve an overall testing event score of satisfactory performance for two consecutive testing events or two out of three consecutive testing events is unsuccessful performance.
§ 493.827 Standard; Mycology.
(a) Failure to attain an overall testing event score of at least 80 percent is unsatisfactory performance.
(b) Failure to participate in a testing event is unsatisfactory performance and results in a score of 0 for the testing event. Consideration may be given to those laboratories failing to participate in a testing event only if—
(1) Patient testing was suspended during the time frame allotted for testing and reporting proficiency testing results;
(2) The laboratory notifies the inspecting agency and the proficiency testing program within the time frame for submitting proficiency testing results of the suspension of patient testing and the circumstances associated with failure to perform tests on proficiency testing samples; and
(3) The laboratory participated in the previous two proficiency testing events.
(c) Failure to return proficiency testing results to the proficiency testing program within the time frame specified by the program is unsatisfactory performance and results in a score of 0 for the testing event.
(d)
(1) For any unsatisfactory testing event for reasons other than a failure to participate, the laboratory must undertake appropriate training and employ the technical assistance necessary to correct problems associated with a proficiency testing failure.
(2) Remedial action must be taken and documented, and the documentation must be maintained by the laboratory for two years from the date of participation in the proficiency testing event.
(e) Failure to achieve an overall testing event score of satisfactory performance for two consecutive testing events or two out of three consecutive testing events is unsuccessful performance.
§ 493.829 Standard; Parasitology.
(a) Failure to attain an overall testing event score of at least 80 percent is unsatisfactory performance.
(b) Failure to participate in a testing event is unsatisfactory performance and results in a score of 0 for the testing event. Consideration may be given to those laboratories failing to participate in a testing event only if—
(1) Patient testing was suspended during the time frame allotted for testing and reporting proficiency testing results;
(2) The laboratory notifies the inspecting agency and the proficiency testing program within the time frame for submitting proficiency testing results of the suspension of patient testing and the circumstances associated with failure to perform tests on proficiency testing samples; and
(3) The laboratory participated in the previous two proficiency testing events.
(c) Failure to return proficiency testing results to the proficiency testing program within the time frame specified by the program is unsatisfactory performance and results in a score of 0 for the testing event.
(d)
(1) For any unsatisfactory testing event for reasons other than a failure to participate, the laboratory must undertake appropriate training and employ the technical assistance necessary to correct problems associated with a proficiency testing failure.
(2) Remedial action must be taken and documented, and the documentation must be maintained by the laboratory for two years from the date of participation in the proficiency testing event.
(e) Failure to achieve an overall testing event score of satisfactory performance for two consecutive testing events or two out of three consecutive testing events is unsuccessful performance.
§ 493.831 Standard; Virology.
(a) Failure to attain an overall testing event score of at least 80 percent is unsatisfactory performance.
(b) Failure to participate in a testing event is unsatisfactory performance and results in a score of 0 for the testing event. Consideration may be given to those laboratories failing to participate in a testing event only if—
(1) Patient testing was suspended during the time frame allotted for testing and reporting proficiency testing results;
(2) The laboratory notifies the inspecting agency and the proficiency testing program within the time frame for submitting proficiency testing results of the suspension of patient testing and the circumstances associated with failure to perform tests on proficiency testing samples; and
(3) The laboratory participated in the previous two proficiency testing events.
(c) Failure to return proficiency testing results to the proficiency testing program within the time frame specified by the program is unsatisfactory performance and results in a score of 0 for the testing event.
(d)
(1) For any unsatisfactory testing event for reasons other than a failure to participate, the laboratory must undertake appropriate training and employ the technical assistance necessary to correct problems associated with a proficiency testing failure.
(2) For any unsatisfactory testing events, remedial action must be taken and documented, and the documentation must be maintained by the laboratory for two years from the date of participation in the proficiency testing event.
(e) Failure to achieve an overall testing event score of satisfactory performance for two consecutive testing events or two out of three consecutive testing events is unsuccessful performance.
§ 493.833 Condition: Diagnostic immunology.
The specialty of diagnostic immunology includes for purposes of proficiency testing the subspecialties of syphilis serology and general immunology.
§ 493.835 Standard; Syphilis serology.
(a) Failure to attain an overall testing event score of at least 80 percent is unsatisfactory performance.
(b) Failure to participate in a testing event is unsatisfactory performance and results in a score of 0 for the testing event. Consideration may be given to those laboratories failing to participate in a testing event only if—
(1) Patient testing was suspended during the time frame allotted for testing and reporting proficiency testing results;
(2) The laboratory notifies the inspecting agency and the proficiency testing program within the time frame for submitting proficiency testing results of the suspension of patient testing and the circumstances associated with failure to perform tests on proficiency testing samples; and
(3) The laboratory participated in the previous two proficiency testing events.
(c) Failure to return proficiency testing results to the proficiency testing program within the time frame specified by the program is unsatisfactory performance and results in a score of 0 for the testing event.
(d)
(1) For any unsatisfactory testing event for reasons other than a failure to participate, the laboratory must undertake appropriate training and employ the technical assistance necessary to correct problems associated with a proficiency testing failure.
(2) For any unacceptable testing event score, remedial action must be taken and documented, and the documentation must be maintained by the laboratory for two years from the date of participation in the proficiency testing event.
(e) Failure to achieve an overall testing event score of satisfactory performance for two consecutive testing events or two out of three consecutive testing events is unsuccessful performance.
§ 493.837 Standard; General immunology.
(a) Failure to attain a score of at least 80 percent of acceptable responses for each analyte in each testing event is unsatisfactory analyte performance for the testing event.
(b) Failure to attain an overall testing event score of at least 80 percent is unsatisfactory performance.
(c) Failure to participate in a testing event is unsatisfactory performance and results in a score of 0 for the testing event. Consideration may be given to those laboratories failing to participate in a testing event only if—
(1) Patient testing was suspended during the time frame allotted for testing and reporting proficiency testing results;
(2) The laboratory notifies the inspecting agency and the proficiency testing program within the time frame for submitting proficiency testing results of the suspension of patient testing and the circumstances associated with failure to perform tests on proficiency testing samples; and
(3) The laboratory participated in the previous two proficiency testing events.
(d) Failure to return proficiency testing results to the proficiency testing program within the time frame specified by the program is unsatisfactory performance and results in a score of 0 for the testing event.
(e)
(1) For any unsatisfactory analyte or test performance or testing event for reasons other than a failure to participate, the laboratory must undertake appropriate training and employ the technical assistance necessary to correct problems associated with a proficiency testing failure.
(2) For any unacceptable analyte or testing event score, remedial action must be taken and documented, and the documentation must be maintained by the laboratory for two years from the date of participation in the proficiency testing event.
(f) Failure to achieve satisfactory performance for the same analyte or test in two consecutive testing events or two out of three consecutive testing events is unsuccessful performance.
(g) Failure to achieve an overall testing event score of satisfactory performance for two consecutive testing events or two out of three consecutive testing events is unsuccessful performance.
§ 493.839 Condition: Chemistry.
The specialty of chemistry includes for the purposes of proficiency testing the subspecialties of routine chemistry, endocrinology, and toxicology.
§ 493.841 Standard; Routine chemistry.
(a) Failure to attain a score of at least 80 percent of acceptable responses for each analyte in each testing event is unsatisfactory analyte performance for the testing event.
(b) Failure to attain an overall testing event score of at least 80 percent is unsatisfactory performance.
(c) Failure to participate in a testing event is unsatisfactory performance and results in a score of 0 for the testing event. Consideration may be given to those laboratories failing to participate in a testing event only if—
(1) Patient testing was suspended during the time frame allotted for testing and reporting proficiency testing results;
(2) The laboratory notifies the inspecting agency and the proficiency testing program within the time frame for submitting proficiency testing results of the suspension of patient testing and the circumstances associated with failure to perform tests on proficiency testing samples; and
(3) The laboratory participated in the previous two proficiency testing events.
(d) Failure to return proficiency testing results to the proficiency testing program within the time frame specified by the program is unsatisfactory performance and results in a score of 0 for the testing event.
(e)
(1) For any unsatisfactory analyte or test performance or testing event for reasons other than a failure to participate, the laboratory must undertake appropriate training and employ the technical assistance necessary to correct problems associated with a proficiency testing failure.
(2) For any unacceptable analyte or testing event score, remedial action must be taken and documented, and the documentation must be maintained by the laboratory for two years from the date of participation in the proficiency testing event.
(f) Failure to achieve satisfactory performance for the same analyte or test in two consecutive testing events or two out of three consecutive testing events is unsuccessful performance.
(g) Failure to achieve an overall testing event score of satisfactory performance for two consecutive testing events or two out of three consecutive testing events is unsuccessful performance.
§ 493.843 Standard; Endocrinology.
(a) Failure to attain a score of at least 80 percent of acceptable responses for each analyte in each testing event is unsatisfactory analyte performance for the testing event.
(b) Failure to attain an overall testing event score of at least 80 percent is unsatisfactory performance.
(c) Failure to participate in a testing event is unsatisfactory performance and results in a score of 0 for the testing event. Consideration may be given to those laboratories failing to participate in a testing event only if—
(1) Patient testing was suspended during the time frame allotted for testing and reporting proficiency testing results;
(2) The laboratory notifies the inspecting agency and the proficiency testing program within the time frame for submitting proficiency testing results of the suspension of patient testing and the circumstances associated with failure to perform tests on proficiency testing samples; and
(3) The laboratory participated in the previous two proficiency testing events.
(d) Failure to return proficiency testing results to the proficiency testing program within the time frame specified by the program is unsatisfactory performance and results in a score of 0 for the testing event.
(e)
(1) For any unsatisfactory analyte or test performance or testing event for reasons other than a failure to participate, the laboratory must undertake appropriate training and employ the technical assistance necessary to correct problems associated with a proficiency testing failure.
(2) For any unacceptable analyte or testing event score, remedial action must be taken and documented, and the documentation must be maintained by the laboratory for two years from the date of participation in the proficiency testing event.
(f) Failure to achieve satisfactory performance for the same analyte or test in two consecutive testing events or two out of three consecutive testing events is unsuccessful performance.
(g) Failure to achieve an overall testing event score of satisfactory performance for two consecutive testing events or two out of three consecutive testing events is unsuccessful performance.
§ 493.845 Standard; Toxicology.
(a) Failure to attain a score of at least 80 percent of acceptable responses for each analyte in each testing event is unsatisfactory analyte performance for the testing event.
(b) Failure to attain an overall testing event score of at least 80 percent is unsatisfactory performance.
(c) Failure to participate in a testing event is unsatisfactory performance and results in a score of 0 for the testing event. Consideration may be given to those laboratories failing to participate in a testing event only if—
(1) Patient testing was suspended during the time frame allotted for testing and reporting proficiency testing results;
(2) The laboratory notifies the inspecting agency and the proficiency testing program within the time frame for submitting proficiency testing results of the suspension of patient testing and the circumstances associated with failure to perform tests on proficiency testing samples; and
(3) The laboratory participated in the previous two proficiency testing events.
(d) Failure to return proficiency testing results to the proficiency testing program within the time frame specified by the program is unsatisfactory performance and results in a score of 0 for the testing event.
(e)
(1) For any unsatisfactory analyte or test performance or testing event for reasons other than a failure to participate, the laboratory must undertake appropriate training and employ the technical assistance necessary to correct problems associated with a proficiency testing failure.
(2) For any unacceptable analyte or testing event score, remedial action must be taken and documented, and the documentation must be maintained by the laboratory for two years from the date of participation in the proficiency testing event.
(f) Failure to achieve satisfactory performance for the same analyte or test in two consecutive testing events or two out of three consecutive testing events is unsuccessful performance.
(g) Failure to achieve an overall testing event score of satisfactory performance for two consecutive testing events or two out of three consecutive testing events is unsuccessful performance.
§ 493.849 Condition: Hematology.
The specialty of hematology, for the purpose of proficiency testing, is not subdivided into subspecialties of testing.
§ 493.851 Standard; Hematology.
(a) Failure to attain a score of at least 80 percent of acceptable responses for each analyte in each testing event is unsatisfactory analyte performance for the testing event.
(b) Failure to attain an overall testing event score of at least 80 percent is unsatisfactory performance.
(c) Failure to participate in a testing event is unsatisfactory performance and results in a score of 0 for the testing event. Consideration may be given to those laboratories failing to participate in a testing event only if—
(1) Patient testing was suspended during the time frame allotted for testing and reporting proficiency testing results;
(2) The laboratory notifies the inspecting agency and the proficiency testing program within the time frame for submitting proficiency testing results of the suspension of patient testing and the circumstances associated with failure to perform tests on proficiency testing samples; and
(3) The laboratory participated in the previous two proficiency testing events.
(d) Failure to return proficiency testing results to the proficiency testing program within the time frame specified by the program is unsatisfactory performance and results in a score of 0 for the testing event.
(e)
(1) For any unsatisfactory analyte or test performance or testing event for reasons other than a failure to participate, the laboratory must undertake appropriate training and employ the technical assistance necessary to correct problems associated with a proficiency testing failure.
(2) For any unacceptable analyte or testing event score, remedial action must be taken and documented, and the documentation must be maintained by the laboratory for two years from the date of participation in the proficiency testing event.
(f) Failure to achieve satisfactory performance for the same analyte in two consecutive events or two out of three consecutive testing events is unsuccessful performance.
(g) Failure to achieve an overall testing event score of satisfactory performance for two consecutive testing events or two out of three consecutive testing events is unsuccessful performance.
§ 493.853 Condition: Pathology.
The specialty of pathology includes, for purposes of proficiency testing, the subspecialty of cytology limited to gynecologic examinations.
§ 493.855 Standard; Cytology: gynecologic examinations.
To participate successfully in a cytology proficiency testing program for gynecologic examinations (Pap smears), the laboratory must meet the requirements of paragraphs (a) through (c) of this section.
(a) The laboratory must ensure that each individual engaged in the examination of gynecologic preparations is enrolled in a proficiency testing program approved by CMS by January 1, 1995, if available in the State in which he or she is employed. The laboratory must ensure that each individual is tested at least once per year and obtains a passing score. To ensure this annual testing of individuals, an announced or unannounced testing event will be conducted on-site in each laboratory at least once each year. Laboratories will be notified of the time of each announced on-site testing event at least 30 days prior to each event. Additional testing events will be conducted as necessary in each State or region for the purpose of testing individuals who miss the on-site testing event and for retesting individuals as described in paragraph (b) of this section.
(b) The laboratory must ensure that each individual participates in an annual testing event that involves the examination of a 10-slide test set as described in § 493.945. Individuals who fail this testing event are retested with another 10-slide test set as described in paragraphs (b)(1) and (b)(2) of this section. Individuals who fail this second test are subsequently retested with a 20-slide test set as described in paragraphs (b)(2) and (b)(3) of this section. Individuals are given not more than 2 hours to complete a 10-slide test and not more than 4 hours to complete a 20-slide test. Unexcused failure to appear by an individual for a retest will result in test failure with resulting remediation and limitations on slide examinations as specified in (b)(1), (b)(2), and (b)(3) of this section.
(1) An individual is determined to have failed the annual testing event if he or she scores less than 90 percent on a 10-slide test set. For an individual who fails an annual proficiency testing event, the laboratory must schedule a retesting event which must take place not more than 45 days after receipt of the notification of failure.
(2) An individual is determined to have failed the second testing event if he or she scores less than 90 percent on a 10-slide test set. For an individual who fails a second testing event, the laboratory must provide him or her with documented, remedial training and education in the area of failure, and must assure that all gynecologic slides evaluated subsequent to the notice of failure are reexamined until the individual is again retested with a 20-slide test set and scores at least 90 percent. Reexamination of slides must be documented.
(3) An individual is determined to have failed the third testing event if he or she scores less than 90 percent on a 20-slide test set. An individual who fails the third testing event must cease examining gynecologic slide preparations immediately upon notification of test failure and may not resume examining gynecologic slides until the laboratory assures that the individual obtains at least 35 hours of documented, formally structured, continuing education in diagnostic cytopathology that focuses on the examination of gynecologic preparations, and until he or she is retested with a 20-slide test set and scores at least 90 percent.
(c) If a laboratory fails to ensure that individuals are tested or those who fail a testing event are retested, or fails to take required remedial actions as described in paragraphs (b)(1), (b)(2) or (b)(3) of this section, CMS will initiate intermediate sanctions or limit the laboratory's certificate to exclude gynecologic cytology testing under CLIA, and, if applicable, suspend the laboratory's Medicare and Medicaid payments for gynecologic cytology testing in accordance with subpart R of this part.
[57 FR 7146, Feb. 28, 1992, as amended at 58 FR 5228, Jan. 19, 1993; 59 FR 62609, Dec. 6, 1994]
§ 493.857 Condition: Immunohematology.
The specialty of immunohematology includes four subspecialties for the purposes of proficiency testing: ABO group and D (Rho) typing; unexpected antibody detection; compatibility testing; and antibody identification.
§ 493.859 Standard; ABO group and D (Rho) typing.
(a) Failure to attain a score of at least 100 percent of acceptable responses for each analyte or test in each testing event is unsatisfactory analyte performance for the testing event.
(b) Failure to attain an overall testing event score of at least 100 percent is unsatisfactory performance.
(c) Failure to participate in a testing event is unsatisfactory performance and results in a score of 0 for the testing event. Consideration may be given to those laboratories failing to participate in a testing event only if—
(1) Patient testing was suspended during the time frame allotted for testing and reporting proficiency testing results;
(2) The laboratory notifies the inspecting agency and the proficiency testing program within the time frame for submitting proficiency testing results of the suspension of patient testing and the circumstances associated with failure to perform tests on proficiency testing samples; and
(3) The laboratory participated in the previous two proficiency testing events.
(d) Failure to return proficiency testing results to the proficiency testing program within the time frame specified by the program is unsatisfactory performance and results in a score of 0 for the testing event.
(e)
(1) For any unsatisfactory testing event for reasons other than a failure to participate, the laboratory must undertake appropriate training and employ the technical assistance necessary to correct problems associated with a proficiency testing failure.
(2) For any unacceptable analyte or unsatisfactory testing event score, remedial action must be taken and documented, and the documentation must be maintained by the laboratory for two years from the date of participation in the proficiency testing event.
(f) Failure to achieve satisfactory performance for the same analyte in two consecutive testing events or two out of three consecutive testing events is unsuccessful performance.
(g) Failure to achieve an overall testing event score of satisfactory for two consecutive testing events or two out of three consecutive testing events is unsuccessful performance.
§ 493.861 Standard; Unexpected antibody detection.
(a) Failure to attain an overall testing event score of at least 80 percent is unsatisfactory performance.
(b) Failure to participate in a testing event is unsatisfactory performance and results in a score of 0 for the testing event. Consideration may be given to those laboratories failing to participate in a testing event only if -
(1) Patient testing was suspended during the time frame allotted for testing and reporting proficiency testing results;
(2) The laboratory notifies the inspecting agency and the proficiency testing program within the time frame for submitting proficiency testing results of the suspension of patient testing and the circumstances associated with failure to perform tests on proficiency testing samples; and
(3) The laboratory participated in the previous two proficiency testing events.
(c) Failure to return proficiency testing results to the proficiency testing program within the time frame specified by the program is unsatisfactory performance and results in a score of 0 for the testing event.
(d)
(1) For any unsatisfactory testing event for reasons other than a failure to participate, the laboratory must undertake appropriate training and employ the technical assistance necessary to correct problems associated with a proficiency testing failure.
(2) For any unsatisfactory testing event score, remedial action must be taken and documented, and the documentation must be maintained by the laboratory for two years from the date of participation in the proficiency testing event.
(e) Failure to achieve an overall testing event score of satisfactory for two consecutive testing events or two out of three consecutive testing events is unsuccessful performance.
§ 493.863 Standard; Compatibility testing.
(a) Failure to attain an overall testing event score of at least 100 percent is unsatisfactory performance.
(b) Failure to participate in a testing event is unsatisfactory performance and results in a score of 0 for the testing event. Consideration may be given to those laboratories failing to participate in a testing event only if—
(1) Patient testing was suspended during the time frame allotted for testing and reporting proficiency testing results;
(2) The laboratory notifies the inspecting agency and the proficiency testing program within the time frame for submitting proficiency testing results of the suspension of patient testing and the circumstances associated with failure to perform tests on proficiency testing samples; and
(3) The laboratory participated in the previous two proficiency testing events.
(c) Failure to return proficiency testing results to the proficiency testing program within the time frame specified by the program is unsatisfactory performance and results in a score of 0 for the testing event.
(d)
(1) For any unsatisfactory testing event for reasons other than a failure to participate, the laboratory must undertake appropriate training and employ the technical assistance necessary to correct problems associated with a proficiency testing failure.
(2) For any unsatisfactory testing event score, remedial action must be taken and documented, and the documentation must be maintained by the laboratory for two years from the date of participation in the proficiency testing event.
(e) Failure to achieve an overall testing event score of satisfactory for two consecutive testing events or two out of three consecutive testing events is unsuccessful performance.
§ 493.865 Standard; Antibody identification.
(a) Failure to attain an overall testing event score of at least 80 percent is unsatisfactory performance.
(b) Failure to participate in a testing event is unsatisfactory performance and results in a score of 0 for the testing event. Consideration may be given to those laboratories failing to participate in a testing event only if—
(1) Patient testing was suspended during the time frame allotted for testing and reporting proficiency testing results;
(2) The laboratory notifies the inspecting agency and the proficiency testing program within the time frame for submitting proficiency testing results of the suspension of patient testing and the circumstances associated with failure to perform tests on proficiency testing samples; and
(3) The laboratory participated in the previous two proficiency testing events.
(c) Failure to return proficiency testing results to the proficiency testing program within the time frame specified by the program is unsatisfactory performance and results in a score of 0 for the testing event.
(d)
(1) For any unsatisfactory testing event for reasons other than a failure to participate, the laboratory must undertake appropriate training and employ the technical assistance necessary to correct problems associated with a proficiency testing failure.
(2) For any unsatisfactory testing event score, remedial action must be taken and documented, and the documentation must be maintained by the laboratory for two years from the date of participation in the proficiency testing event.
(e) Failure to identify the same antibody in two consecutive or two out of three consecutive testing events is unsuccessful performance.
(f) Failure to achieve an overall testing event score of satisfactory for two consecutive testing events or two out of three consecutive testing events is unsuccessful performance.
Subpart I - Proficiency Testing Programs for Nonwaived Testing
Source:
57 FR 7151, Feb. 28, 1992, unless otherwise noted.
§ 493.901 Approval of proficiency testing programs.
In order for a proficiency testing program to receive HHS approval, the program must be offered by a private nonprofit organization or a Federal or State agency, or entity acting as a designated agent for the State. An organization, Federal, or State program seeking approval or reapproval for its program for the next calendar year must submit an application providing the required information by July 1 of the current year. The organization, Federal, or State program must provide technical assistance to laboratories seeking to qualify under the program, and must, for each specialty, subspecialty, and analyte or test for which it provides testing -
(a) Assure the quality of test samples, appropriately evaluate and score the testing results, and identify performance problems in a timely manner;
(b) Demonstrate to HHS that it has -
(1) The technical ability required to -
(i) Prepare or purchase samples from manufacturers who prepare the samples in conformance with the appropriate good manufacturing practices required in 21 CFR parts 606, 640, and 820; and
(ii) Distribute the samples, using rigorous quality control to assure that samples mimic actual patient specimens when possible and that samples are homogeneous, except for specific subspecialties such as cytology, and will be stable within the time frame for analysis by proficiency testing participants;
(2) A scientifically defensible process for determining the correct result for each challenge offered by the program;
(3) A program of sufficient annual challenge and with the frequency specified in §§ 493.909 through 493.959 to establish that a laboratory has met minimum performance requirements;
(4) The resources needed to provide Statewide or nationwide reports to regulatory agencies on individual's performance for gynecologic cytology and on individual laboratory performance on testing events, cumulative reports and scores for each laboratory or individual, and reports of specific laboratory failures using grading criteria acceptable to HHS. These reports must be provided to HHS on a timely basis when requested;
(5) Provisions to include on each proficiency testing program report form used by the laboratory to record testing event results, an attestation statement that proficiency testing samples were tested in the same manner as patient specimens with a signature block to be completed by the individual performing the test as well as by the laboratory director;
(6) A mechanism for notifying participants of the PT shipping schedule and for participants to notify the proficiency testing program within three days of the expected date of receipt of the shipment that samples have not arrived or are unacceptable for testing. The program must have provisions for replacement of samples that are lost in transit or are received in a condition that is unacceptable for testing; and
(7) A process to resolve technical, administrative, and scientific problems about program operations;
(c) Meet the specific criteria for proficiency testing programs listed by specialty, subspecialty, and analyte or test contained in §§ 493.901 through 493.959 for initial approval and thereafter provide HHS, on an annual basis, with the information necessary to assure that the proficiency testing program meets the criteria required for approval; and
(d) Comply with all applicable packaging, shipment, and notification requirements of 42 CFR part 72.
[57 FR 7151, Feb. 28, 1992, as amended at 58 FR 5228, Jan. 19, 1993]
§ 493.903 Administrative responsibilities.
The proficiency testing program must -
(a)
(1) Provide HHS or its designees and participating laboratories with an electronic or a hard copy, or both, of reports of proficiency testing results and all scores for each laboratory's performance in a format as required by and approved by CMS for each CLIA-certified specialty, subspecialty, and analyte or test within 60 days after the date by which the laboratory must report proficiency testing results to the proficiency testing program.
(2) Provide HHS with reports of PT results and scores of individual performance in cytology and provide copies of reports to participating individuals, and to all laboratories that employ the individuals, within 15 working days of the testing event;
(b) Furnish to HHS cumulative reports on an individual laboratory's performance and aggregate data on CLIA-certified laboratories for the purpose of establishing a system to make the proficiency testing program's results available, on a reasonable basis, upon request of any person, and include such explanatory information as may be appropriate to assist in the interpretation of the proficiency testing program's results;
(c) Provide HHS with additional information and data upon request and submit such information necessary for HHS to conduct an annual evaluation to determine whether the proficiency testing program continues to meet the requirements of §§ 493.901 through 493.959;
(d) Maintain records of laboratories' performance for a period of five years or such time as may be necessary for any legal proceedings; and
(e) Provide HHS with an annual report and, if needed, an interim report which identifies any previously unrecognized sources of variability in kits, instruments, methods, or PT samples, which adversely affect the programs' ability to evaluate laboratory performance.
[57 FR 7151, Feb. 28, 1992, as amended at 58 FR 5228, Jan. 19, 1993]
§ 493.905 Nonapproved proficiency testing programs.
If a proficiency testing program is determined by HHS to fail to meet any criteria contained in §§ 493.901 through 493.959 for approval of the proficiency testing program, CMS will notify the program and the program must notify all laboratories enrolled of the nonapproval and the reasons for nonapproval within 30 days of the notification.
Proficiency Testing Programs by Specialty and Subspecialty
§ 493.909 Microbiology.
The subspecialties under the specialty of microbiology for which a program may offer proficiency testing are bacteriology, mycobacteriology, mycology, parasitology and virology. Specific criteria for these subspecialties are found at §§ 493.911 through 493.919.
§ 493.911 Bacteriology.
(a) Types of services offered by laboratories. In bacteriology, for proficiency testing purposes, there are five types of laboratories:
(1) Those that interpret Gram stains or perform primary inoculation, or both; and refer cultures to another laboratory appropriately certified for the subspecialty of bacteriology for identification;
(2) Those that use direct antigen techniques to detect an organism and may also interpret Gram stains or perform primary inoculation, or perform any combination of these;
(3) Those that, in addition to interpreting Gram stains, performing primary inoculations, and using direct antigen tests, also isolate and identify aerobic bacteria from throat, urine, cervical, or urethral discharge specimens to the genus level and may also perform antimicrobial susceptibility tests on selected isolated microorganisms;
(4) Those that perform the services in paragraph (a)(3) of this section and also isolate and identify aerobic bacteria from any source to the species level and may also perform antimicrobial susceptibility tests; and
(5) Those that perform the services in paragraph (a)(4) of this section and also isolate and identify anaerobic bacteria from any source.
(b) Program content and frequency of challenge. To be approved for proficiency testing for bacteriology, the annual program must provide a minimum of five samples per testing event. There must be at least three testing events at approximately equal intervals per year. The samples may be provided to the laboratory through mailed shipments or, at HHS' option, may be provided to HHS or its designee for on-site testing. For the types of laboratories specified in paragraph (a) of this section, an annual program must include samples that contain organisms that are representative of the six major groups of bacteria: anaerobes, Enterobacteriaceae, gram-positive bacilli, gram-positive cocci, gram-negative cocci, and miscellaneous gram-negative bacteria, as appropriate. The specific organisms included in the samples may vary from year to year. The annual program must include samples for bacterial antigen detection, bacterial isolation and identification, Gram stain, and antimicrobial susceptibility testing.
(1) An approved program must furnish HHS with a description of samples that it plans to include in its annual program no later than six months before each calendar year. At least 50 percent of the samples must be mixtures of the principal organism and appropriate normal flora. The program must include other important emerging pathogens (as determined by HHS) and either organisms commonly occurring in patient specimens or opportunistic pathogens. The program must include the following two types of samples; each type of sample must meet the 50 percent mixed culture criterion:
(i) Samples that require laboratories to report only organisms that the testing laboratory considers to be a principal pathogen that is clearly responsible for a described illness (excluding immuno-compromised patients). The program determines the reportable isolates, including antimicrobial susceptibility for any designated isolate; and
(ii) Samples that require laboratories to report all organisms present. Samples must contain multiple organisms frequently found in specimens such as urine, blood, abscesses, and aspirates where multiple isolates are clearly significant or where specimens are derived from immuno-compromised patients. The program determines the reportable isolates.
(2) An approved program may vary over time. For example, the types of organisms that might be included in an approved program over time are -
Anaerobes:
Bacteroides fragilis group
Clostridium perfringens
Peptostreptococcus anaerobius
Enterobacteriaceae
Citrobacter freundii
Enterobacter aerogenes
Escherichia coli
Klebsiella pneumoniae
Proteus mirabilis
Salmonella typhimurium
Serratia marcescens
Shigella sonnei
Yersinia enterocolitica
Gram-positive bacilli:
Listeria monocytogenes
Corynebacterium species CDC Group JK
Gram-positive cocci:
Staphylococcus aureus
Streptococcus Group A
Streptococcus Group B
Streptococcus Group D (S. bovis and enterococcus)
Streptococcus pneumoniae
Gram-negative cocci:
Branhamella catarrhalis
Neisseria gonorrhoeae
Neisseria meningitidis
Miscellaneous Gram-negative bacteria:
Campylobacter jejuni
Haemophilis influenza, Type B
Pseudomonas aeruginosa
(3) For antimicrobial susceptibility testing, the program must provide at least one sample per testing event that includes gram-positive or gram-negative strains that have a predetermined pattern of sensitivity or resistance to the common antimicrobial agents.
(c) Evaluation of a laboratory's performance. HHS approves only those programs that assess the accuracy of a laboratory's responses in accordance with paragraphs (c) (1) through (7) of this section.
(1) The program determines staining characteristics to be interpreted by Gram stain. The program determines the reportable bacteria to be detected by direct antigen techniques or isolation. To determine the accuracy of a laboratory's response for Gram stain interpretation, direct antigen detection, identification, or antimicrobial susceptibility testing, the program must compare the laboratory's response for each sample with the response which reflects agreement of either 80 percent of ten or more referee laboratories or 80 percent or more of all participating laboratories.
(2) To evaluate a laboratory's response for a particular sample, the program must determine a laboratory's type of service in accordance with paragraph (a) of this section. A laboratory must isolate and identify the organisms to the same extent it performs these procedures on patient specimens. A laboratory's performance will be evaluated on the basis of its final answer, for example, a laboratory specified in paragraph (a)(3) of this section will be evaluated on the basis of the average of its scores for paragraphs (c)(3) through (c)(6) as determined in paragraph (c)(7) of this section.
(3) Since laboratories may incorrectly report the presence of organisms in addition to the correctly identified principal organism(s), the grading system must provide a means of deducting credit for additional erroneous organisms that are reported. Therefore, the total number of correct responses for organism isolation and identification submitted by the laboratory divided by the number of organisms present plus the number of incorrect organisms reported by the laboratory must be multiplied by 100 to establish a score for each sample in each testing event. For example, if a sample contained one principal organism and the laboratory reported it correctly but reported the presence of an additional organism, which was not considered reportable, the sample grade would be 1/(1 + 1) × 100 = 50 percent.
(4) For antimicrobial susceptibility testing, a laboratory must indicate which drugs are routinely included in its test panel when testing patient samples. A laboratory's performance will be evaluated for only those antibiotics for which service is offered. A correct response for each antibiotic will be determined as described in § 493.911(c) (1) using criteria such as the guidelines established by the National Committee for Clinical Laboratory Standards. Grading is based on the number of correct susceptibility responses reported by the laboratory divided by the actual number of correct susceptibility responses determined by the program, multiplied by 100. For example, if a laboratory offers susceptibility testing for Enterobacteriaceae using amikacin, cephalothin, and tobramycin, and the organism in the proficiency testing sample is an Enterobacteriaceae, and the laboratory reports correct responses for two of three antimicrobial agents, the laboratory's grade would be 2/3 × 100 = 67 percent.
(5) The performance criterion for qualitative antigen tests is the presence or absence of the bacterial antigen. The score for antigen tests is the number of correct responses divided by the number of samples to be tested for the antigen, multiplied by 100.
(6) The performance criteria for Gram stain is staining reaction, i.e., gram positive or gram negative. The score for Gram stain is the number of correct responses divided by the number of challenges to be tested, multiplied by 100.
(7) The score for a testing event in bacteriology is the average of the scores determined under paragraphs (c)(3) through (c)(6) of this section kbased on the type of service offered by the laboratory.
[57 FR 7151, Feb. 28, 1992, as amended at 58 FR 5228, Jan. 19, 1993; 68 FR 3702, Jan. 24, 2003]
§ 493.913 Mycobacteriology.
(a) Types of services offered by laboratories. In mycobacteriology, there are five types of laboratories for proficiency testing purposes:
(1) Those that interpret acid-fast stains and refer specimen to another laboratory appropriately certified in the subspecialty of mycobacteriology;
(2) Those that interpret acid-fast stains, perform primary inoculation, and refer cultures to another laboratory appropriately certified in the subspecialty of mycobacteriology for identification;
(3) Those that interpret acid-fast stains, isolate and perform identification and/or antimycobacterial susceptibility of Mycobacterium tuberculosis, but refer other mycobacteria species to another laboratory appropriately certified in the subspecialty of mycobacteriology for identification and/or susceptibility tests;
(4) Those that interpret acid-fast stains, isolate and identify all mycobacteria to the extent required for correct clinical diagnosis, but refer antimycobacterial susceptibility tests to another laboratory appropriately certified in the subspecialty of mycobacteriology; and
(5) Those that interpret acid-fast stains, isolate and identify all mycobacteria to the extent required for correct clinical diagnosis, and perform antimycobacterial susceptibility tests on the organisms isolated.
(b) Program content and frequency of challenge. To be approved for proficiency testing for mycobacteriology, the annual program must provide a minimum of five samples per testing event. There must be at least two testing events per year. The samples may be provided through mailed shipments or, at HHS' option, provided to HHS or its designee for on-site testing events. For types of laboratories specified in paragraphs (a)(1) and (a) (3) through (5) of this section, an annual program must include samples that contain species that are representative of the 5 major groups (complexes) of mycobacteria encountered in human specimens. The specific mycobacteria included in the samples may vary from year to year.
(1) An approved program must furnish HHS and its agents with a description of samples that it plans to include in its annual program no later than six months before each calendar year. At least 50 percent of the samples must be mixtures of the principal mycobacteria and appropriate normal flora. The program must include mycobacteria commonly occurring in patient specimens and other important emerging mycobacteria (as determined by HHS). The program determines the reportable isolates and correct responses for antimycobacterial susceptibility for any designated isolate.
(2) An approved program may vary over time. For example, the types of mycobacteria that might be included in an approved program over time are -
TB
Mycobacterium tuberculosis
Mycobacterium bovis
Group I
Mycobacterium kansasii
Group II
Mycobacterium szulgai
Group III
Mycobacterium avium-intracellulare
Mycobacterium terrae
Group IV
Mycobacterium fortuitum
(3) For antimycobacterial susceptibility testing, the program must provide at least one sample per testing event that includes mycobacterium tuberculosis that has a predetermined pattern of sensitivity or resistance to the common antimycobacterial agents.
(4) For laboratories specified in paragraphs (a)(1) and (a)(2), the program must provide at least five samples per testing event that includes challenges that are acid-fast and challenges which do not contain acid-fast organisms.
(c) Evaluation of a laboratory's performance. HHS approves only those programs that assess the accuracy of a laboratory's response in accordance with paragraphs (c)(1) through (6) of this section.
(1) The program determines the reportable mycobacteria to be detected by acid-fast stain, for isolation and identification, and for antimycobacterial susceptibility. To determine the accuracy of a laboratory's response, the program must compare the laboratory's response for each sample with the response that reflects agreement of either 80 percent of ten or more referee laboratories or 80 percent or more of all participating laboratories.
(2) To evaluate a laboratory's response for a particular sample, the program must determine a laboratory's type of service in accordance with paragraph (a) of this section. A laboratory must interpret acid-fast stains and isolate and identify the organisms to the same extent it performs these procedures on patient specimens. A laboratory's performance will be evaluated on the basis of the average of its scores as determined in paragraph (c)(6) of this section.
(3) Since laboratories may incorrectly report the presence of organisms in addition to the correctly identified principal organism(s), the grading system must provide a means of deducting credit for additional erroneous organisms reported. Therefore, the total number of correct responses submitted by the laboratory divided by the number of organisms present plus the number of incorrect organisms reported by the laboratory must be multiplied by 100 to establish a score for each sample in each testing event. For example, if a sample contained one principal organism and the laboratory reported it correctly but reported the presence of an additional organism, which was not present, the sample grade would be
1 / (1 + 1) × 100 = 50 percent
(4) For antimycobacterial susceptibility testing, a laboratory must indicate which drugs are routinely included in its test panel when testing patient samples. A laboratory's performance will be evaluated for only those antibiotics for which susceptibility testing is routinely performed on patient specimens. A correct response for each antibiotic will be determined as described in § 493.913(c)(1). Grading is based on the number of correct susceptibility responses reported by the laboratory divided by the actual number of correct susceptibility responses as determined by the program, multiplied by 100. For example, if a laboratory offers susceptibility testing using three antimycobacterial agents and the laboratory reports correct response for two of the three antimycobacterial agents, the laboratory's grade would be 2⁄3 × 100 = 67 percent.
(5) The performance criterion for qualitative tests is the presence or absence of acid-fast organisms. The score for acid-fast organism detection is the number of correct responses divided by the number of samples to be tested, multiplied by 100.
(6) The score for a testing event in mycobacteriology is the average of the scores determined under paragraphs (c)(3) through (c)(5) of this section based on the type of service offered by the laboratory.
[57 FR 7151, Feb. 28, 1992, as amended at 58 FR 5228, Jan. 19, 1993; 68 FR 3702, Jan. 24, 2003]
§ 493.915 Mycology.
(a) Types of services offered by laboratories. In mycology, there are four types of laboratories for proficiency testing purposes that may perform different levels of service for yeasts, dimorphic fungi, dermatophytes, and aerobic actinomycetes:
(1) Those that isolate and identify only yeasts and/or dermatophytes to the genus level;
(2) Those that isolate and identify yeasts and/or dermatophytes to the species level;
(3) Those that isolate and perform identification of all organisms to the genus level; and
(4) Those that isolate and perform identification of all organisms to the species level.
(b) Program content and frequency of challenge. To be approved for proficiency testing for mycology, the annual program must provide a minimum of five samples per testing event. There must be at least three testing events at approximately equal intervals per year. The samples may be provided through mailed shipments or, at HHS' option, may be provided to HHS or its designee for on-site testing. An annual program must include samples that contain organisms that are representative of five major groups of fungi: Yeast or yeast-like fungi; dimorphic fungi; dematiaceous fungi; dermatophytes; and saprophytes, including opportunistic fungi. The specific fungi included in the samples may vary from year to year.
(1) An approved program must, before each calendar year, furnish HHS with a description of samples that it plans to include in its annual program no later than six months before each calendar year. At least 50 percent of the samples must be mixtures of the principal organism and appropriate normal background flora. Other important emerging pathogens (as determined by HHS) and organisms commonly occurring in patient specimens must be included periodically in the program.
(2) An approved program may vary over time. As an example, the types of organisms that might be included in an approved program over time are -
Candida albicans
Candida (other species)
Cryptococcus neoformans
Sporothrix schenckii
Exophiala jeanselmei
Fonsecaea pedrosoi
Microsporum sp.
Acremonium sp.
Trichophvton sp.
Aspergillus fumigatus
Nocardia sp.
Blastomyces dermatitidis1
Zygomycetes sp.
1 Note: Provided as a nonviable sample.
(c) Evaluation of a laboratory's performance. HHS approves only those programs that assess the accuracy of a laboratory's response, in accordance with paragraphs (c)(1) through (5) of this section.
(1) The program determines the reportable organisms. To determine the accuracy of a laboratory's response, the program must compare the laboratory's response for each sample with the response that reflects agreement of either 80 percent of ten or more referee laboratories or 80 percent or more of all participating laboratories.
(2) To evaluate a laboratory's response for a particular sample, the program must determine a laboratory's type of service in accordance with paragraph (a) of this section. A laboratory must isolate and identify the organisms to the same extent it performs these procedures on patient specimens.
(3) Since laboratories may incorrectly report the presence of organisms in addition to the correctly identified principal organism(s), the grading system must deduct credit for additional erroneous organisms reported. Therefore, the total number of correct responses submitted by the laboratory divided by the number of organisms present plus the number of incorrect organisms reported by the laboratory must be multiplied by 100 to establish a score for each sample in each shipment or testing event. For example, if a sample contained one principal organism and the laboratory reported it correctly but reported the presence of an additional organism, which was not present, the sample grade would be 1/(1 + 1)x100 = 50 percent.
(4) The score for the antigen tests is the number of correct responses divided by the number of samples to be tested for the antigen, multiplied by 100.
(5) The score for a testing event is the average of the sample scores as determined under paragraph (c)(3) or (c)(4), or both, of this section.
[57 FR 7151, Feb. 28, 1992, as amended at 58 FR 5228, Jan. 19, 1993; 68 FR 3702, Jan. 24, 2003]
§ 493.917 Parasitology.
(a) Types of services offered by laboratories. In parasitology there are two types of laboratories for proficiency testing purposes -
(1) Those that determine the presence or absence of parasites by direct observation (wet mount) and/or pinworm preparations and, if necessary, refer specimens to another laboratory appropriately certified in the subspecialty of parasitology for identification;
(2) Those that identify parasites using concentration preparations and/or permanent stains.
(b) Program content and frequency of challenge. To be approved for proficiency testing in parasitology, a program must provide a minimum of five samples per testing event. There must be at least three testing events at approximately equal intervals per year. The samples may be provided through mailed shipments or, at HHS's option, may be provided to HHS or its designee for on-site testing. An annual program must include samples that contain parasites that are commonly encountered in the United States as well as those recently introduced into the United States. Other important emerging pathogens (as determined by HHS) and parasites commonly occurring in patient specimens must be included periodically in the program.
(1) An approved program must, before each calendar year furnish HHS with a description of samples that it plans to include in its annual program no later than six months before each calendar year. Samples must include both formalinized specimens and PVA (polyvinyl alcohol) fixed specimens as well as blood smears, as appropriate for a particular parasite and stage of the parasite. The majority of samples must contain protozoa or helminths or a combination of parasites. Some samples must be devoid of parasites.
(2) An approved program may vary over time. As an example, the types of parasites that might be included in an approved program over time are -
Enterobius vermicularis
Entamoeba histolytica
Entamoeba coli
Giardia lamblia
Endolimax nana
Dientamoeba fragilis
Iodamoeba butschli
Chilomastix mesnili
Hookworm
Ascaris lumbricoides
Strongyloides stercoralis
Trichuris trichiura
Diphyllobothrium latum
Cryptosporidium sp.
Plasmodium falciparum
(3) For laboratories specified in paragraph (a)(1) of this section, the program must provide at least five samples per testing event that include challenges which contain parasites and challenges that are devoid of parasites.
(c) Evaluation of a laboratory's performance. HHS approves only those programs that assess the accuracy of a laboratory's responses in accordance with paragraphs (c)(1) through (6) of this section.
(1) The program must determine the reportable parasites. It may elect to establish a minimum number of parasites to be identified in samples before they are reported. Parasites found in rare numbers by referee laboratories are not considered in scoring a laboratory's performance; such findings are neutral. To determine the accuracy of a laboratory's response, the program must compare the laboratory's response with the response that reflects agreement of either 80 percent of ten or more referee laboratories or 80 percent or more of all participating laboratories.
(2) To evaluate a laboratory's response for a particular sample, the program must determine a laboratory's type of service in accordance with paragraph (a) of this section. A laboratory must determine the presence or absence of a parasite(s) or concentrate and identify the parasites to the same extent it performs these procedures on patient specimens.
(3) Since laboratories may incorrectly report the presence of parasites in addition to the correctly identified principal parasite(s), the grading system must deduct credit for these additional erroneous parasites reported and not found in rare numbers by the program's referencing process. Therefore, the total number of correct responses submitted by the laboratory divided by the number of parasites present plus the number of incorrect parasites reported by the laboratory must be multiplied by 100 to establish a score for each sample in each testing event. For example, if a sample contained one principal parasite and the laboratory reported it correctly but reported the presence of an additional parasite, which was not present, the sample grade would be
1/(1 + 1) × 100 = 50 percent.
(4) The criterion for acceptable performance for qualitative parasitology examinations is presence or absence of a parasite(s).
(5) The score for parasitology is the number of correct responses divided by the number of samples to be tested, multiplied by 100.
(6) The score for a testing event is the average of the sample scores as determined under paragraphs (c)(3) through (c)(5) of this section.
[57 FR 7151, Feb. 28, 1992, as amended at 68 FR 3702, Jan. 24, 2003]
§ 493.919 Virology.
(a) Types of services offered by laboratories. In virology, there are two types of laboratories for proficiency testing purposes -
(1) Those that only perform tests that directly detect viral antigens or structures, either in cells derived from infected tissues or free in fluid specimens; and
(2) Those that are able to isolate and identify viruses and use direct antigen techniques.
(b) Program content and frequency of challenge. To be approved for proficiency testing in virology, a program must provide a minimum of five samples per testing event. There must be at least three testing events at approximately equal intervals per year. The samples may be provided to the laboratory through mailed shipments or, at HHS's option, may be provided to HHS or its designee for on-site testing. An annual program must include viral species that are the more commonly identified viruses. The specific organisms found in the samples may vary from year to year. The annual program must include samples for viral antigen detection and viral isolation and identification.
(1) An approved program must furnish HHS with a description of samples that it plans to include in its annual program no later than six months before each calendar year. The program must include other important emerging viruses (as determined by HHS) and viruses commonly occurring in patient specimens.
(2) An approved program may vary over time. For example, the types of viruses that might be included in an approved program over time are the more commonly identified viruses such as Herpes simplex, respiratory syncytial virus, adenoviruses, enteroviruses, and cytomegaloviruses.
(c) Evaluation of laboratory's performance. HHS approves only those programs that assess the accuracy of a laboratory's response in accordance with paragraphs (c)(1) through (5) of this section.
(1) The program determines the reportable viruses to be detected by direct antigen techniques or isolated by laboratories that perform viral isolation procedures. To determine the accuracy of a laboratory's response, the program must compare the laboratory's response for each sample with the response that reflects agreement of either 80 percent of ten or more referee laboratories or 80 percent or more of all participating laboratories.
(2) To evaluate a laboratory's response for a particular sample, the program must determine a laboratory's type of service in accordance with paragraph (a) of this section. A laboratory must isolate and identify the viruses to the same extent it performs these procedures on patient specimens.
(3) Since laboratories may incorrectly report the presence of viruses in addition to the correctly identified principal virus, the grading system must provide a means of deducting credit for additional erroneous viruses reported. Therefore, the total number of correct responses determined by virus culture techniques submitted by the laboratory divided by the number of viruses present plus the number of incorrect viruses reported by the laboratory must be multiplied by 100 to establish a score for each sample in each testing event. For example, if a sample contained one principal virus and the laboratory reported it correctly but reported the presence of an additional virus, which was not present, the sample grade would be 1/(1 + 1) × 100 = 50 percent.
(4) The performance criterion for qualitative antigen tests is presence or absence of the viral antigen. The score for the antigen tests is the number of correct responses divided by the number of samples to be tested for the antigen, multiplied by 100.
(5) The score for a testing event is the average of the sample scores as determined under paragraph (c)(3) and (c)(4) of this section.
[57 FR 7151, Feb. 28, 1992, as amended at 68 FR 3702, Jan. 24, 2003]
§ 493.921 Diagnostic immunology.
The subspecialties under the specialty of immunology for which a program may offer proficiency testing are syphilis serology and general immunology. Specific criteria for these subspecialties are found at §§ 493.923 and 493.927.
§ 493.923 Syphilis serology.
(a) Program content and frequency of challenge. To be approved for proficiency testing in syphilis serology, a program must provide a minimum of five samples per testing event. There must be at least three testing events at approximately equal intervals per year. The samples may be provided through mailed shipments or, at HHS' option, may be provided to HHS or its designee for on-site testing. An annual program must include samples that cover the full range of reactivity from highly reactive to non-reactive.
(b) Evaluation of test performance. HHS approves only those programs that assess the accuracy of a laboratory's responses in accordance with paragraphs (b)(1) through (4) of this section.
(1) To determine the accuracy of a laboratory's response for qualitative and quantitative syphilis tests, the program must compare the laboratory's response with the response that reflects agreement of either 80 percent of ten or more referee laboratories or 80 percent or more of all participating laboratories. The proficiency testing program must indicate the minimum concentration, by method, that will be considered as indicating a positive response. The score for a sample in syphilis serology is the average of scores determined under paragraphs (b)(2) and (b)(3) of this section.
(2) For quantitative syphilis tests, the program must determine the correct response for each method by the distance of the response from the target value. After the target value has been established for each response, the appropriateness of the response must be determined by using fixed criteria. The criterion for acceptable performance for quantitative syphilis serology tests is the target value ±1 dilution.
(3) The criterion for acceptable performance for qualitative syphilis serology tests is reactive or nonreactive.
(4) To determine the overall testing event score, the number of correct responses must be averaged using the following formula:

[57 FR 7151, Feb. 28, 1992, as amended at 58 FR 5229, Jan. 19, 1993; 68 FR 3702, Jan. 24, 2003]
§ 493.927 General immunology.
(a) Program content and frequency of challenge. To be approved for proficiency testing for immunology, the annual program must provide a minimum of five samples per testing event. There must be at least three testing events at approximately equal intervals per year. The annual program must provide samples that cover the full range of reactivity from highly reactive to nonreactive. The samples may be provided through mailed shipments or, at HHS' option, may be provided to HHS or its designee for on-site testing.
(b) Challenges per testing event. The minimum number of challenges per testing event the program must provide for each analyte or test procedure is five. Analytes or tests for which laboratory performance is to be evaluated include:
Analyte or Test Procedure
Alpha-l antitrypsin
Alpha-fetoprotein (tumor marker)
Antinuclear antibody
Antistreptolysin O
Anti-human immunodeficiency virus (HIV)
Complement C3
Complement C4
Hepatitis markers (HBsAg, anti-HBc, HBeAg)
IgA
IgG
IgE
IgM
Infectious mononucleosis
Rheumatoid factor
Rubella
(c) Evaluation of a laboratory's analyte or test performance. HHS approves only those programs that assess the accuracy of a laboratory's responses in accordance with paragraphs (c)(1) through (5) of this section.
(1) To determine the accuracy of a laboratory's response for quantitative and qualitative immunology tests or analytes, the program must compare the laboratory's response for each analyte with the response that reflects agreement of either 80 percent of ten or more referee laboratories or 80 percent or more of all participating laboratories. The proficiency testing program must indicate the minimum concentration that will be considered as indicating a positive response. The score for a sample in general immunology is either the score determined under paragraph (c)(2) or (3) of this section.
(2) For quantitative immunology analytes or tests, the program must determine the correct response for each analyte by the distance of the response from the target value. After the target value has been established for each response, the appropriateness of the response must be determined by using either fixed criteria or the number of standard deviations (SDs) the response differs from the target value.
Criteria for Acceptable Performance
The criteria for acceptable performance are -
| Analyte or test | Criteria for acceptable performance |
|---|---|
| Alpha-1 antitrypsin | Target value ±3 SD. |
| Alpha-fetoprotein (tumor marker) | Target value ±3 SD. |
| Antinuclear antibody | Target value ±2 dilutions or positive or negative. |
| Antistreptolysin O | Target value ±2 dilution or positive or negative. |
| Anti-Human Immunodeficiency virus | Reactive or nonreactive. |
| Complement C3 | Target value ±3 SD. |
| Complement C4 | Target value ±3 SD. |
| Hepatitis (HBsAg, anti-HBc, HBeAg) | Reactive (positive) or nonreactive (negative). |
| IgA | Target value ±3 SD. |
| IgE | Target value ±3 SD. |
| IgG | Target value ±25%. |
| IgM | Target value ±3 SD. |
| Infectious mononucleosis | Target value ±2 dilutions or positive or negative. |
| Rheumatoid factor | Target value ±2 dilutions or positive or negative. |
| Rubella | Target value ±2 dilutions or immune or nonimmune or positive or negative. |
(3) The criterion for acceptable performance for qualitative general immunology tests is positive or negative.
(4) To determine the analyte testing event score, the number of acceptable analyte responses must be averaged using the following formula:

(5) To determine the overall testing event score, the number of correct responses for all analytes must be averaged using the following formula:
[57 FR 7151, Feb. 28, 1992, as amended at 58 FR 5229, Jan. 19, 1993; 68 FR 3702, Jan. 24, 2003]
§ 493.929 Chemistry.
The subspecialties under the specialty of chemistry for which a proficiency testing program may offer proficiency testing are routine chemistry, endocrinology, and toxicology. Specific criteria for these subspecialties are listed in §§ 493.931 through 493.939.
§ 493.931 Routine chemistry.
(a) Program content and frequency of challenge. To be approved for proficiency testing for routine chemistry, a program must provide a minimum of five samples per testing event. There must be at least three testing events at approximately equal intervals per year. The annual program must provide samples that cover the clinically relevant range of values that would be expected in patient specimens. The specimens may be provided through mailed shipments or, at HHS' option, may be provided to HHS or its designee for on-site testing.
(b) Challenges per testing event. The minimum number of challenges per testing event a program must provide for each analyte or test procedure listed below is five serum, plasma or blood samples.
Analyte or Test Procedure
Alanine aminotransferase (ALT/SGPT)
Albumin
Alkaline phosphatase
Amylase
Aspartate aminotransferase (AST/SGOT)
Bilirubin, total
Blood gas (pH, pO2, and pCO2)
Calcium, total
Chloride
Cholesterol, total
Cholesterol, high density lipoprotein
Creatine kinase
Creatine kinase, isoenzymes
Creatinine
Glucose (Excluding measurements on devices cleared by FDA for home use)
Iron, total
Lactate dehydrogenase (LDH)
LDH isoenzymes
Magnesium
Potassium
Sodium
Total Protein
Triglycerides
Urea Nitrogen
Uric Acid
(c) Evaluation of a laboratory's analyte or test performance. HHS approves only those programs that assess the accuracy of a laboratory's responses in accordance with paragraphs (c)(1) through (5) of this section.
(1) To determine the accuracy of a laboratory's response for qualitative and quantitative chemistry tests or analytes, the program must compare the laboratory's response for each analyte with the response that reflects agreement of either 80 percent of ten or more referee laboratories or 80 percent or more of all participating laboratories. The score for a sample in routine chemistry is either the score determined under paragraph (c)(2) or (3) of this section.
(2) For quantitative chemistry tests or analytes, the program must determine the correct response for each analyte by the distance of the response from the target value. After the target value has been established for each response, the appropriateness of the response must be determined by using either fixed criteria based on the percentage difference from the target value or the number of standard deviations (SDs) the response differs from the target value.
Criteria for Acceptable Performance
The criteria for acceptable performance are -
| Analyte or test | Criteria for acceptable performance |
|---|---|
| Alanine aminotransferase (ALT/SGPT) | Target value ±20%. |
| Albumin | Target value ±10%. |
| Alkaline phosphatase | Target value ±30%. |
| Amylase | Target value ±30%. |
| Aspartate aminotransferase (AST/SGOT) | Target value ±20%. |
| Bilirubin, total | Target value ±0.4 mg/dL or ±20% (greater). |
| Blood gas pO2 | Target value ±3 SD. |
| pCO2 | Target value ±5 mm Hg or ±8% (greater). |
| pH | Target value ±0.04. |
| Calcium, total | Target value ±1.0 mg/dL. |
| Chloride | Target value ±5%. |
| Cholesterol, total | Target value ±10%. |
| Cholesterol, high density lipoprotein | Target value ±30%. |
| Creatine kinase | Target value ±30%. |
| Creatine kinase isoenzymes | MB elevated (presence or absence) or Target value ±3SD. |
| Creatinine | Target value ±0.3 mg/dL or ±15% (greater). |
| Glucose (excluding glucose performed on monitoring devices cleared by FDA for home use | Target value ±6 mg/dl or ±10% (greater). |
| Iron, total | Target value ±20%. |
| Lactate dehydrogenase (LDH) | Target value ±20%. |
| LDH isoenzymes | LDH1/LDH2 (+ or −) or Target value ±30%. |
| Magnesium | Target value ±25%. |
| Potassium | Target value ±0.5 mmol/L. |
| Sodium | Target value ±4 mmol/L. |
| Total Protein | Target value ±10%. |
| Triglycerides | Target value ±25%. |
| Urea nitrogen | Target value ±2 mg/dL or ±9% (greater). |
| Uric acid | Target value ±17%. |
(3) The criterion for acceptable performance for qualitative routine chemistry tests is positive or negative.
(4) To determine the analyte testing event score, the number of acceptable analyte responses must be averaged using the following formula:
(5) To determine the overall testing event score, the number of correct responses for all analytes must be averaged using the following formula:
[57 FR 7151, Feb. 28, 1992, as amended at 68 FR 3702, Jan. 24, 2003]
§ 493.933 Endocrinology.
(a) Program content and frequency of challenge. To be approved for proficiency testing for endocrinology, a program must provide a minimum of five samples per testing event. There must be at least three testing events at approximately equal intervals per year. The annual program must provide samples that cover the clinically relevant range of values that would be expected in patient specimens. The samples may be provided through mailed shipments or, at HHS' option, may be provided to HHS or its designee for on-site testing.
(b) Challenges per testing event. The minimum number of challenges per testing event a program must provide for each analyte or test procedure is five serum, plasma, blood, or urine samples.
Analyte or Test
Cortisol
Free Thyroxine
Human Chorionic gonadotropin (excluding urine pregnancy tests done by visual color comparison categorized as waived tests)
T3 Uptake
Triiodothyronine
Thyroid-stimulating hormone
Thyroxine
(c) Evaluation of a laboratory's analyte or test performance. HHS approves only those programs that assess the accuracy of a laboratory's responses in accordance with paragraphs (c)(1) through (5) of this section.
(1) To determine the accuracy of a laboratory's response for qualitative and quantitative endocrinology tests or analytes, a program must compare the laboratory's response for each analyte with the response that reflects agreement of either 80 percent of ten or more referee laboratories or 80 percent or more of all participating laboratories. The score for a sample in endocrinology is either the score determined under paragraph (c)(2) or (c)(3) of this section.
(2) For quantitative endocrinology tests or analytes, the program must determine the correct response for each analyte by the distance of the response from the target value. After the target value has been established for each response, the appropriateness of the response must be determined by using either fixed criteria based on the percentage difference from the target value or the number of standard deviations (SDs) the response differs from the target value.
Criteria for Acceptable Performance
The criteria for acceptable performance are -
| Analyte or test | Criteria for acceptable performance |
|---|---|
| Cortisol | Target value ±25%. |
| Free Thyroxine | Target value ±3 SD. |
| Human Chorionic Gonadotropin (excluding urine pregnancy tests done by visual color comparison categorized as waived tests) | Target value ±3 SD positive or negative. |
| T3 Uptake | Target value ±3 SD. |
| Triiodothyronine | Target value ±3 SD. |
| Thyroid-stimulating hormone | Target value ±3 SD. |
| Thyroxine | Target value ±20% or 1.0 mcg/dL (greater). |
(3) The criterion for acceptable performance for qualitative endocrinology tests is positive or negative.
(4) To determine the analyte testing event score, the number of acceptable analyte responses must be averaged using the following formula:
(5) To determine the overall testing event score, the number of correct responses for all analytes must be averaged using the following formula:
[57 FR 7151, Feb. 28, 1992, as amended at 58 FR 5229, Jan. 19, 1993; 68 FR 3702, Jan. 24, 2003]
§ 493.937 Toxicology.
(a) Program content and frequency of challenge. To be approved for proficiency testing for toxicology, the annual program must provide a minimum of five samples per testing event. There must be at least three testing events at approximately equal intervals per year. The annual program must provide samples that cover the clinically relevant range of values that would be expected in specimens of patients on drug therapy and that cover the level of clinical significance for the particular drug. The samples may be provided through mailed shipments or, at HHS' option, may be provided to HHS or its designee for on-site testing.
(b) Challenges per testing event. The minimum number of challenges per testing event a program must provide for each analyte or test procedure is five serum, plasma, or blood samples.
Analyte or Test Procedure
- Alcohol (blood)
- Blood lead
- Carbamazepine
- Digoxin
- Ethosuximide
- Gentamicin
- Lithium
- Phenobarbital
- Phenytoin
- Primidone
- Procainamide
- (and metabolite)
- Quinidine
- Theophylline
- Tobramycin
- Valproic Acid
(c) Evaluation of a laboratory's analyte or test performance. HHS approves only those programs that assess the accuracy of a laboratory's responses in accordance with paragraphs (c)(1) through (4) of this section.
(1) To determine the accuracy of a laboratory's responses for quantitative toxicology tests or analytes, the program must compare the laboratory's response for each analyte with the response that reflects agreement of either 80 percent of ten or more referee laboratories or 80 percent or more of all participating laboratories. The score for a sample in toxicology is the score determined under paragraph (c)(2) of this section.
(2) For quantitative toxicology tests or analytes, the program must determine the correct response for each analyte by the distance of the response from the target value. After the target value has been established for each response, the appropriateness of the response must be determined by using fixed criteria based on the percentage difference from the target value
Criteria for Acceptable Performance
The criteria for acceptable performance are:
| Analyte or test | Criteria for acceptable performance |
|---|---|
| Alcohol, blood | Target Value ±25%. |
| Blood lead | Target Value ±10% or 4 mcg/dL (greater). |
| Carbamazepine | Target Value ±25%. |
| Digoxin | Target Value ±20% or ±0.2 ng/mL (greater). |
| Ethosuximide | Target Value ±20%. |
| Gentamicin | Target Value ±25%. |
| Lithium | Target Value ±0.3 mmol/L or ±20% (greater). |
| Phenobarbital | Target Value ±20% |
| Phenytoin | Target Value ±25%. |
| Primidone | Target Value ±25%. |
| Procainamide (and metabolite) | Target Value ±25%. |
| Quinidine | Target Value ±25%. |
| Tobramycin | Target Value ±25%. |
| Theophylline | Target Value ±25%. |
| Valproic Acid | Target Value ±25%. |
(3) To determine the analyte testing event score, the number of acceptable analyte responses must be averaged using the following formula:
(4) To determine the overall testing event score, the number of correct responses for all analytes must be averaged using the following formula:
[57 FR 7151, Feb. 28, 1992, as amended at 58 FR 5229, Jan. 19, 1993; 68 FR 3702, Jan. 24, 2003]
§ 493.941 Hematology (including routine hematology and coagulation).
(a) Program content and frequency of challenge. To be approved for proficiency testing for hematology, a program must provide a minimum of five samples per testing event. There must be at least three testing events at approximately equal intervals per year. The annual program must provide samples that cover the full range of values that would be expected in patient specimens. The samples may be provided through mailed shipments or, at HHS' option, may be provided to HHS and or its designee for on-site testing.
(b) Challenges per testing event. The minimum number of challenges per testing event a program must provide for each analyte or test procedure is five.
Analyte or Test Procedure
Cell identification or white blood cell differential
Erythrocyte count
Hematocrit (excluding spun microhematocrit)
Hemoglobin
Leukocyte count
Platelet count
Fibrinogen
Partial thromboplastin time
Prothrombin time
(1) An approved program for cell identification may vary over time. The types of cells that might be included in an approved program over time are -
Neutrophilic granulocytes
Eosinophilic granulocytes
Basophilic granulocytes
Lymphocytes
Monocytes
Major red and white blood cell abnormalities
Immature red and white blood cells
(2) White blood cell differentials should be limited to the percentage distribution of cellular elements listed above.
(c) Evaluation of a laboratory's analyte or test performance. HHS approves only those programs that assess the accuracy of a laboratory's responses in accordance with paragraphs (c) (1) through (5) of this section.
(1) To determine the accuracy of a laboratory's responses for qualitative and quantitative hematology tests or analytes, the program must compare the laboratory's response for each analyte with the response that reflects agreement of either 80 percent of ten or more referee laboratories or 80 percent or more of all participating laboratories. The score for a sample in hematology is either the score determined under paragraph (c) (2) or (3) of this section.
(2) For quantitative hematology tests or analytes, the program must determine the correct response for each analyte by the distance of the response from the target value. After the target value has been established for each response, the appropriateness of the response is determined using either fixed criteria based on the percentage difference from the target value or the number of standard deviations (SDs) the response differs from the target value.
Criteria for Acceptable Performance
The criteria for acceptable performance are:
| Analyte or test | Criteria for acceptable performance |
|---|---|
| Cell identification | 90% or greater consensus on identification. |
| White blood cell differential | Target ±3SD based on the percentage of different types of white blood cells in the samples. |
| Erythrocyte count | Target ±6%. |
| Hematocrit (Excluding spun hematocrits) | Target ±6%. |
| Hemoglobin | Target ±7%. |
| Leukocyte count | Target ±15%. |
| Platelet count | Target ±25%. |
| Fibrinogen | Target ±20%. |
| Partial thromboplastin time | Target ±15%. |
| Prothrombin time | Target ±15%. |
(3) The criterion for acceptable performance for the qualitative hematology test is correct cell identification.
(4) To determine the analyte testing event score, the number of acceptable analyte responses must be averaged using the following formula:
(5) To determine the overall testing event score, the number of correct responses for all analytes must be averaged using the following formula:
[57 FR 7151, Feb. 28, 1992, as amended at 58 FR 5229, Jan. 19, 1993; 68 FR 3702, Jan. 24, 2003]
§ 493.945 Cytology; gynecologic examinations.
(a) Program content and frequency of challenge.
(1) To be approved for proficiency testing for gynecologic examinations (Pap smears) in cytology, a program must provide test sets composed of 10- and 20-glass slides. Proficiency testing programs may obtain slides for test sets from cytology laboratories, provided the slides have been retained by the laboratory for the required period specified in §§ 493.1105(a)(7)(i)(A) and 493.1274(f)(2). If slide preparations are still subject to retention by the laboratory, they may be loaned to a proficiency testing program if the program provides the laboratory with documentation of the loan of the slides and ensures that slides loaned to it are retrievable upon request. Each test set must include at least one slide representing each of the response categories described in paragraph (b)(3)(ii)(A) of this section, and test sets should be comparable so that equitable testing is achieved within and between proficiency testing providers.
(2) To be approved for proficiency testing in gynecologic cytology, a program must provide announced and unannounced on-site testing for each individual at least once per year and must provide an initial retesting event for each individual within 45 days after notification of test failure and subsequent retesting events within 45 days after completion of remedial action described in § 493.855.
(b) Evaluation of an individual's performance. HHS approves only those programs that assess the accuracy of each individual's responses on both 10- and 20-slide test sets in which the slides have been referenced as specified in paragraph (b)(1) of this section.
(1) To determine the accuracy of an individual's response on a particular challenge (slide), the program must compare the individual's response for each slide preparation with the response that reflects the predetermined consensus agreement or confirmation on the diagnostic category, as described in the table in paragraph (b)(3)(ii)(A) of this section. For all slide preparations, a 100% consensus agreement among a minimum of three physicians certified in anatomic pathology is required. In addition, for premalignant and malignant slide preparations, confirmation by tissue biopsy is required either by comparison of the reported biopsy results or reevaluation of biopsy slide material by a physician certified in anatomic pathology.
(2) An individual qualified as a technical supervisor under § 493.1449 (b) or (k) who routinely interprets gynecologic slide preparations only after they have been examined by a cytotechnologist can either be tested using a test set that has been screened by a cytotechnologist in the same laboratory or using a test set that has not been screened. A technical supervisor who screens and interprets slide preparations that have not been previously examined must be tested using a test set that has not been previously screened.
(3) The criteria for acceptable performance are determined by using the scoring system in paragraphs (b)(3) (i) and (ii) of this section.
(i) Each slide set must contain 10 or 20 slides with point values established for each slide preparation based on the significance of the relationship of the interpretation of the slide to a clinical condition and whether the participant in the testing event is a cytotechnologist qualified under § 493.1469 or § 493.1483 or functioning as a technical supervisor in cytology qualified under § 493.1449 (b) or (k) of this part.
(ii) The scoring system rewards or penalizes the participants in proportion to the distance of their answers from the correct response or target diagnosis and the penalty or reward is weighted in proportion to the severity of the lesion.
(A) The four response categories for reporting proficiency testing results and their descriptions are as follows:
| Category | Description |
|---|---|
| A | Unsatisfactory for diagnosis due to: |
| (1) Scant cellularity. | |
| (2) Air drying. | |
| (3) Obscuring material (blood, inflammatory cells, or lubricant). | |
| B | Normal or Benign Changes—includes: |
| (1) Normal, negative or within normal limits. | |
| (2) Infection other than Human Papillomavirus (HPV) (e.g., Trichomonas vaginalis, changes or morphology consistent with Candida spp., Actinomyces spp. or Herpes simplex virus). | |
| (3) Reactive and reparative changes (e.g., inflammation, effects of chemotherapy or radiation). | |
| C | Low Grade Squamous Intraepithelial Lesion—includes: |
| (1) Cellular changes associated with HPV. | |
| (2) Mild dysplasia/CIN-1. | |
| D | High Grade Lesion and Carcinoma—includes: |
| (1) High grade squamous intraepithelial lesions which include moderate dysplasia/CIN-2 and severe dysplasia/carcinoma in-situ/CIN-3. | |
| (2) Squamous cell carcinoma. | |
| (3) Adenocarcinoma and other malignant neoplasms. |
(B) In accordance with the criteria for the scoring system, the charts in paragraphs (b)(3)(ii)(C) and (D) of this section, for technical supervisors and cytotechnologists, respectively, provide a maximum of 10 points for a correct response and a maximum of minus five (−5) points for an incorrect response on a 10-slide test set. For example, if the correct response on a slide is “high grade squamous intraepithelial lesion” (category “D” on the scoring system chart) and an examinee calls it “normal or negative” (category “B” on the scoring system chart), then the examinee's point value on that slide is calculated as minus five (−5). Each slide is scored individually in the same manner. The individual's score for the testing event is determined by adding the point value achieved for each slide preparation, dividing by the total points for the testing event and multiplying by 100.
(C) Criteria for scoring system for a 10-slide test set. (See table at (b)(3)(ii)(A) of this section for a description of the response categories.) For technical supervisors qualified under § 493.1449(b) or (k):
| Examinee's response: | A | B | C | D |
|---|---|---|---|---|
| Correct response category: | ||||
| A | 10 | 0 | 0 | 0 |
| B | 5 | 10 | 0 | 0 |
| C | 5 | 0 | 10 | 5 |
| D | 0 | −5 | 5 | 10 |
(D) Criteria for scoring system for a 10-slide test set. (See table at paragraph (b)(3)(ii)(A) of this section for a description of the response categories.) For cytotechnologists qualified under § 493.1469 or § 493.1483:
| Examinee's response: | A | B | C | D |
|---|---|---|---|---|
| Correct response category: | ||||
| A | 10 | 0 | 5 | 5 |
| B | 5 | 10 | 5 | 5 |
| C | 5 | 0 | 10 | 10 |
| D | 0 | −5 | 10 | 10 |
(E) In accordance with the criteria for the scoring system, the charts in paragraphs (b)(3)(ii)(F) and (G) of this section, for technical supervisors and cytotechnologists, respectively, provide maximums of 5 points for a correct response and minus ten (−10) points for an incorrect response on a 20-slide test set.
(F) Criteria for scoring system for a 20-slide test set. (See table at paragraph (b)(3)(ii)(A) of this section for a description of the response categories.) For technical supervisors qualified under § 493.1449(b) or (k):
| Examinee's response: | A | B | C | D |
|---|---|---|---|---|
| Correct response category: | ||||
| A | 5 | 0 | 0 | 0 |
| B | 2.5 | 5 | 0 | 0 |
| C | 2.5 | 0 | 5 | 2.5 |
| D | 0 | −10 | 2.5 | 5 |
(G) Criteria for scoring system for a 20-slide test set. (See table at (b)(3)(ii)(A) of this section for a description of the response categories.) For cytotechnologists qualified under § 493.1469 or § 493.1483:
| Examinee's response: | A | B | C | D |
|---|---|---|---|---|
| Correct response category: | ||||
| A | 5 | 0 | 2.5 | 2.5 |
| B | 2.5 | 5 | 2.5 | 2.5 |
| C | 2.5 | 0 | 5 | 5 |
| D | 0 | −10 | 5 | 5 |
[57 FR 7151, Feb. 28, 1992, as amended at 58 FR 5229, Jan. 19, 1993; 68 FR 3702, Jan. 24, 2003]
§ 493.959 Immunohematology.
(a) Types of services offered by laboratories. In immunohematology, there are four types of laboratories for proficiency testing purposes -
(1) Those that perform ABO group and/or D (Rho) typing;
(2) Those that perform ABO group and/or D (Rho) typing, and unexpected antibody detection;
(3) Those that in addition to paragraph (a)(2) of this section perform compatibility testing; and
(4) Those that perform in addition to paragraph (a)(3) of this section antibody identification.
(b) Program content and frequency of challenge. To be approved for proficiency testing for immunohematology, a program must provide a minimum of five samples per testing event. There must be at least three testing events at approximately equal intervals per year. The annual program must provide samples that cover the full range of interpretation that would be expected in patient specimens. The samples may be provided through mailed shipments or, at HHS' option, may be provided to HHS or its designee for on-site testing.
(c) Challenges per testing event. The minimum number of challenges per testing event a program must provide for each analyte or test procedure is five.
Analyte or Test Procedure
ABO group (excluding subgroups)
D (Rho) typing
Unexpected antibody detection
Compatibility testing
Antibody identification
(d) Evaluation of a laboratory's analyte or test performance. HHS approves only those programs that assess the accuracy of a laboratory's response in accordance with paragraphs (d)(1) through (5) of this section.
(1) To determine the accuracy of a laboratory's response, a program must compare the laboratory's response for each analyte with the response that reflects agreement of either 100 percent of ten or more referee laboratories or 95 percent or more of all participating laboratories except for unexpected antibody detection and antibody identification. To determine the accuracy of a laboratory's response for unexpected antibody detection and antibody identification, a program must compare the laboratory's response for each analyte with the response that reflects agreement of either 95 percent of ten or more referee laboratories or 95 percent or more of all participating laboratories. The score for a sample in immunohematology is either the score determined under paragraph (d)(2) or (3) of this section.
(2) Criteria for acceptable performance. The criteria for acceptable performance are -
| Analyte or test | Criteria for acceptable performance |
|---|---|
| ABO group | 100% accuracy. |
| D (Rho) typing | 100% accuracy. |
| Unexpected antibody detection | 80% accuracy. |
| Compatibility testing | 100% accuracy. |
| Antibody identification | 80% accuracy. |
(3) The criterion for acceptable performance for qualitative immunohematology tests is positive or negative.
(4) To determine the analyte testing event score, the number of acceptable analyte responses must be averaged using the following formula:
(5) To determine the overall testing event score, the number of correct responses for all analytes must be averaged using the following formula:
Subpart J - Facility Administration for Nonwaived Testing
Source:
68 FR 3703, Jan. 24, 2003, unless otherwise noted.
§ 493.1100 Condition: Facility administration.
Each laboratory that performs nonwaived testing must meet the applicable requirements under §§ 493.1101 through 493.1105, unless HHS approves a procedure that provides equivalent quality testing as specified in Appendix C of the State Operations Manual (CMS Pub. 7).
(a) Reporting of SARS-CoV-2 test results. During the Public Health Emergency, as defined in § 400.200 of this chapter, each laboratory that performs a test that is intended to detect SARS-CoV-2 or to diagnose a possible case of COVID-19 (hereinafter referred to as a “SARS-CoV-2 test”) must report SARS-CoV-2 test results to the Secretary in such form and manner, and at such timing and frequency, as the Secretary may prescribe.
(b) [Reserved]
[68 FR 3703, Jan. 24, 2003, as amended at 85 FR 54873, Sept. 2, 2020]
§ 493.1101 Standard: Facilities.
(a) The laboratory must be constructed, arranged, and maintained to ensure the following:
(1) The space, ventilation, and utilities necessary for conducting all phases of the testing process.
(2) Contamination of patient specimens, equipment, instruments, reagents, materials, and supplies is minimized.
(3) Molecular amplification procedures that are not contained in closed systems have a uni-directional workflow. This must include separate areas for specimen preparation, amplification and product detection, and, as applicable, reagent preparation.
(b) The laboratory must have appropriate and sufficient equipment, instruments, reagents, materials, and supplies for the type and volume of testing it performs.
(c) The laboratory must be in compliance with applicable Federal, State, and local laboratory requirements.
(d) Safety procedures must be established, accessible, and observed to ensure protection from physical, chemical, biochemical, and electrical hazards, and biohazardous materials.
(e) Records and, as applicable, slides, blocks, and tissues must be maintained and stored under conditions that ensure proper preservation.
§ 493.1103 Standard: Requirements for transfusion services.
A facility that provides transfusion services must meet all of the requirements of this section and document all transfusion-related activities.
(a) Arrangement for services. The facility must have a transfusion service agreement reviewed and approved by the responsible party(ies) that govern the procurement, transfer, and availability of blood and blood products.
(b) Provision of testing. The facility must provide prompt ABO grouping, D(Rho) typing, unexpected antibody detection, compatibility testing, and laboratory investigation of transfusion reactions on a continuous basis through a CLIA-certified laboratory or a laboratory meeting equivalent requirements as determined by CMS.
(c) Blood and blood products storage and distribution.
(1) If a facility stores or maintains blood or blood products for transfusion outside of a monitored refrigerator, the facility must ensure the storage conditions, including temperature, are appropriate to prevent deterioration of the blood or blood product.
(2) The facility must establish and follow policies to ensure positive identification of a blood or blood product beneficiary.
(d) Investigation of transfusion reactions. The facility must have procedures for preventing transfusion reactions and when necessary, promptly identify, investigate, and report blood and blood product transfusion reactions to the laboratory and, as appropriate, to Federal and State authorities.
§ 493.1105 Standard: Retention requirements.
(a) The laboratory must retain its records and, as applicable, slides, blocks, and tissues as follows:
(1) Test requisitions and authorizations. Retain records of test requisitions and test authorizations, including the patient's chart or medical record if used as the test requisition or authorization, for at least 2 years.
(2) Test procedures. Retain a copy of each test procedure for at least 2 years after a procedure has been discontinued. Each test procedure must include the dates of initial use and discontinuance.
(3) Analytic systems records. Retain quality control and patient test records (including instrument printouts, if applicable) and records documenting all analytic systems activities specified in §§ 493.1252 through 493.1289 for at least 2 years. In addition, retain the following:
(i) Records of test system performance specifications that the laboratory establishes or verifies under § 493.1253 for the period of time the laboratory uses the test system but no less than 2 years.
(ii) Immunohematology records, blood and blood product records, and transfusion records as specified in 21 CFR 606.160(b)(3)(ii), (b)(3)(iv), (b)(3)(v) and (d).
(4) Proficiency testing records. Retain all proficiency testing records for at least 2 years.
(5) Quality system assessment records. Retain all laboratory quality systems assessment records for at least 2 years.
(6) Test reports. Retain or be able to retrieve a copy of the original report (including final, preliminary, and corrected reports) at least 2 years after the date of reporting. In addition, retain the following:
(i) Immunohematology reports as specified in 21 CFR 606.160(d).
(ii) Pathology test reports for at least 10 years after the date of reporting.
(7) Slide, block, and tissue retention —
(i) Slides.
(A) Retain cytology slide preparations for at least 5 years from the date of examination (see § 493.1274(f) for proficiency testing exception).
(B) Retain histopathology slides for at least 10 years from the date of examination.
(ii) Blocks. Retain pathology specimen blocks for at least 2 years from the date of examination.
(iii) Tissue. Preserve remnants of tissue for pathology examination until a diagnosis is made on the specimen.
(b) If the laboratory ceases operation, the laboratory must make provisions to ensure that all records and, as applicable, slides, blocks, and tissue are retained and available for the time frames specified in this section.
[68 FR 3703, Jan. 24, 2003; 68 FR 50723, Aug. 22, 2003]
Subpart K - Quality System for Nonwaived Testing
Source:
68 FR 3703, Jan. 24, 2003, unless otherwise noted.
§ 493.1200 Introduction.
(a) Each laboratory that performs nonwaived testing must establish and maintain written policies and procedures that implement and monitor a quality system for all phases of the total testing process (that is, preanalytic, analytic, and postanalytic) as well as general laboratory systems.
(b) The laboratory's quality systems must include a quality assessment component that ensures continuous improvement of the laboratory's performance and services through ongoing monitoring that identifies, evaluates and resolves problems.
(c) The various components of the laboratory's quality system are used to meet the requirements in this part and must be appropriate for the specialties and subspecialties of testing the laboratory performs, services it offers, and clients it serves.
[68 FR 3703, Jan. 24, 2003; 68 FR 50724, Aug. 22, 2003]
§ 493.1201 Condition: Bacteriology.
If the laboratory provides services in the subspecialty of Bacteriology, the laboratory must meet the requirements specified in §§ 493.1230 through 493.1256, § 493.1261, and §§ 493.1281 through 493.1299.
§ 493.1202 Condition: Mycobacteriology.
If the laboratory provides services in the subspecialty of Mycobacteriology, the laboratory must meet the requirements specified in §§ 493.1230 through 493.1256, § 493.1262, and §§ 493.1281 through 493.1299.
§ 493.1203 Condition: Mycology.
If the laboratory provides services in the subspecialty of Mycology, the laboratory must meet the requirements specified in §§ 493.1230 through 493.1256, § 493.1263, and §§ 493.1281 through 493.1299.
§ 493.1204 Condition: Parasitology.
If the laboratory provides services in the subspecialty of Parasitology, the laboratory must meet the requirements specified in §§ 493.1230 through 493.1256, § 493.1264, and §§ 493.1281 through 493.1299.
§ 493.1205 Condition: Virology.
If the laboratory provides services in the subspecialty of Virology, the laboratory must meet the requirements specified in §§ 493.1230 through 493.1256, § 493.1265, and §§ 493.1281 through 493.1299.
§ 493.1207 Condition: Syphilis serology.
If the laboratory provides services in the subspecialty of Syphilis serology, the laboratory must meet the requirements specified in §§ 493.1230 through 493.1256, and §§ 493.1281 through 493.1299.
§ 493.1208 Condition: General immunology.
If the laboratory provides services in the subspecialty of General immunology, the laboratory must meet the requirements specified in §§ 493.1230 through 493.1256, and §§ 493.1281 through 493.1299.
[68 FR 3703, Jan. 24, 2003; 68 FR 50724, Aug. 22, 2003]
§ 493.1210 Condition: Routine chemistry.
If the laboratory provides services in the subspecialty of Routine chemistry, the laboratory must meet the requirements specified in §§ 493.1230 through 493.1256, § 493.1267, and §§ 493.1281 through 493.1299.
§ 493.1211 Condition: Urinalysis.
If the laboratory provides services in the subspecialty of Urinalysis, the laboratory must meet the requirements specified in §§ 493.1230 through 493.1256, and §§ 493.1281 through 493.1299.
§ 493.1212 Condition: Endocrinology.
If the laboratory provides services in the subspecialty of Endocrinology, the laboratory must meet the requirements specified in §§ 493.1230 through 493.1256, and §§ 493.1281 through 493.1299.
§ 493.1213 Condition: Toxicology.
If the laboratory provides services in the subspecialty of Toxicology, the laboratory must meet the requirements specified in §§ 493.1230 through 493.1256, and §§ 493.1281 through 493.1299.
§ 493.1215 Condition: Hematology.
If the laboratory provides services in the specialty of Hematology, the laboratory must meet the requirements specified in §§ 493.1230 through 493.1256, § 493.1269, and §§ 493.1281 through 493.1299.
§ 493.1217 Condition: Immunohematology.
If the laboratory provides services in the specialty of Immunohematology, the laboratory must meet the requirements specified in §§ 493.1230 through 493.1256, § 493.1271, and §§ 493.1281 through 493.1299.
§ 493.1219 Condition: Histopathology.
If the laboratory provides services in the subspecialty of Histopathology, the laboratory must meet the requirements specified in §§ 493.1230 through 493.1256, § 493.1273, and §§ 493.1281 through 493.1299.
§ 493.1220 Condition: Oral pathology.
If the laboratory provides services in the subspecialty of Oral pathology, the laboratory must meet the requirements specified in §§ 493.1230 through 493.1256, and §§ 493.1281 through 493.1299.
§ 493.1221 Condition: Cytology.
If the laboratory provides services in the subspecialty of Cytology, the laboratory must meet the requirements specified in §§ 493.1230 through 493.1256, § 493.1274, and §§ 493.1281 through 493.1299.
§ 493.1225 Condition: Clinical cytogenetics.
If the laboratory provides services in the specialty of Clinical cytogenetics, the laboratory must meet the requirements specified in §§ 493.1230 through 493.1256, § 493.1276, and §§ 493.1281 through 493.1299.
§ 493.1226 Condition: Radiobioassay.
If the laboratory provides services in the specialty of Radiobioassay, the laboratory must meet the requirements specified in §§ 493.1230 through 493.1256, and §§ 493.1281 through 493.1299.
§ 493.1227 Condition: Histocompatibility.
If the laboratory provides services in the specialty of Histocompatibility, the laboratory must meet the requirements specified in §§ 493.1230 through 493.1256, § 493.1278, and §§ 493.1281 through 493.1299.
General Laboratory Systems
§ 493.1230 Condition: General laboratory systems.
Each laboratory that performs nonwaived testing must meet the applicable general laboratory systems requirements in §§ 493.1231 through 493.1236, unless HHS approves a procedure, specified in Appendix C of the State Operations Manual (CMS Pub. 7), that provides equivalent quality testing. The laboratory must monitor and evaluate the overall quality of the general laboratory systems and correct identified problems as specified in § 493.1239 for each specialty and subspecialty of testing performed.
§ 493.1231 Standard: Confidentiality of patient information.
The laboratory must ensure confidentiality of patient information throughout all phases of the total testing process that are under the laboratory's control.
§ 493.1232 Standard: Specimen identification and integrity.
The laboratory must establish and follow written policies and procedures that ensure positive identification and optimum integrity of a patient's specimen from the time of collection or receipt of the specimen through completion of testing and reporting of results.
§ 493.1233 Standard: Complaint investigations.
The laboratory must have a system in place to ensure that it documents all complaints and problems reported to the laboratory. The laboratory must conduct investigations of complaints, when appropriate.
§ 493.1234 Standard: Communications.
The laboratory must have a system in place to identify and document problems that occur as a result of a breakdown in communication between the laboratory and an authorized person who orders or receives test results.
[68 FR 3703, Jan. 24, 2003; 68 FR 50724, Aug. 22, 2003]
§ 493.1235 Standard: Personnel competency assessment policies.
As specified in the personnel requirements in subpart M, the laboratory must establish and follow written policies and procedures to assess employee and, if applicable, consultant competency.
§ 493.1236 Standard: Evaluation of proficiency testing performance.
(a) The laboratory must review and evaluate the results obtained on proficiency testing performed as specified in subpart H of this part.
(b) The laboratory must verify the accuracy of the following:
(1) Any analyte or subspecialty without analytes listed in subpart I of this part that is not evaluated or scored by a CMS-approved proficiency testing program.
(2) Any analyte, specialty or subspecialty assigned a proficiency testing score that does not reflect laboratory test performance (that is, when the proficiency testing program does not obtain the agreement required for scoring as specified in subpart I of this part, or the laboratory receives a zero score for nonparticipation, or late return of results).
(c) At least twice annually, the laboratory must verify the accuracy of the following:
(1) Any test or procedure it performs that is not included in subpart I of this part.
(2) Any test or procedure listed in subpart I of this part for which compatible proficiency testing samples are not offered by a CMS-approved proficiency testing program.
(d) All proficiency testing evaluation and verification activities must be documented.
§ 493.1239 Standard: General laboratory systems quality assessment.
(a) The laboratory must establish and follow written policies and procedures for an ongoing mechanism to monitor, assess, and, when indicated, correct problems identified in the general laboratory systems requirements specified at §§ 493.1231 through 493.1236.
(b) The general laboratory systems quality assessment must include a review of the effectiveness of corrective actions taken to resolve problems, revision of policies and procedures necessary to prevent recurrence of problems, and discussion of general laboratory systems quality assessment reviews with appropriate staff.
(c) The laboratory must document all general laboratory systems quality assessment activities.
[68 FR 3703, Jan. 24, 2003; 68 FR 50724, Aug. 22, 2003]
Preanalytic Systems
§ 493.1240 Condition: Preanalytic systems.
Each laboratory that performs nonwaived testing must meet the applicable preanalytic system(s) requirements in §§ 493.1241 and 493.1242, unless HHS approves a procedure, specified in Appendix C of the State Operations Manual (CMS Pub. 7), that provides equivalent quality testing. The laboratory must monitor and evaluate the overall quality of the preanalytic systems and correct identified problems as specified in § 493.1249 for each specialty and subspecialty of testing performed.
§ 493.1241 Standard: Test request.
(a) The laboratory must have a written or electronic request for patient testing from an authorized person.
(b) The laboratory may accept oral requests for laboratory tests if it solicits a written or electronic authorization within 30 days of the oral request and maintains the authorization or documentation of its efforts to obtain the authorization.
(c) The laboratory must ensure the test requisition solicits the following information:
(1) The name and address or other suitable identifiers of the authorized person requesting the test and, if appropriate, the individual responsible for using the test results, or the name and address of the laboratory submitting the specimen, including, as applicable, a contact person to enable the reporting of imminently life threatening laboratory results or panic or alert values.
(2) The patient's name or unique patient identifier.
(3) The sex and age or date of birth of the patient.
(4) The test(s) to be performed.
(5) The source of the specimen, when appropriate.
(6) The date and, if appropriate, time of specimen collection.
(7) For Pap smears, the patient's last menstrual period, and indication of whether the patient had a previous abnormal report, treatment, or biopsy.
(8) Any additional information relevant and necessary for a specific test to ensure accurate and timely testing and reporting of results, including interpretation, if applicable.
(d) The patient's chart or medical record may be used as the test requisition or authorization but must be available to the laboratory at the time of testing and available to CMS or a CMS agent upon request.
(e) If the laboratory transcribes or enters test requisition or authorization information into a record system or a laboratory information system, the laboratory must ensure the information is transcribed or entered accurately.
§ 493.1242 Standard: Specimen submission, handling, and referral.
(a) The laboratory must establish and follow written policies and procedures for each of the following, if applicable:
(1) Patient preparation.
(2) Specimen collection.
(3) Specimen labeling, including patient name or unique patient identifier and, when appropriate, specimen source.
(4) Specimen storage and preservation.
(5) Conditions for specimen transportation.
(6) Specimen processing.
(7) Specimen acceptability and rejection.
(8) Specimen referral.
(b) The laboratory must document the date and time it receives a specimen.
(c) The laboratory must refer a specimen for testing only to a CLIA-certified laboratory or a laboratory meeting equivalent requirements as determined by CMS.
(d) If the laboratory accepts a referral specimen, written instructions must be available to the laboratory's clients and must include, as appropriate, the information specified in paragraphs (a)(1) through (a)(7) of this section.
§ 493.1249 Standard: Preanalytic systems quality assessment.
(a) The laboratory must establish and follow written policies and procedures for an ongoing mechanism to monitor, assess, and when indicated, correct problems identified in the preanalytic systems specified at §§ 493.1241 through 493.1242.
(b) The preanalytic systems quality assessment must include a review of the effectiveness of corrective actions taken to resolve problems, revision of policies and procedures necessary to prevent recurrence of problems, and discussion of preanalytic systems quality assessment reviews with appropriate staff.
(c) The laboratory must document all preanalytic systems quality assessment activities.
[68 FR 3703, Jan. 24, 2003; 68 FR 3703, Aug. 22, 2003]
Analytic Systems
§ 493.1250 Condition: Analytic systems.
Each laboratory that performs nonwaived testing must meet the applicable analytic systems requirements in §§ 493.1251 through 493.1283, unless HHS approves a procedure, specified in Appendix C of the State Operations Manual (CMS Pub. 7), that provides equivalent quality testing. The laboratory must monitor and evaluate the overall quality of the analytic systems and correct identified problems as specified in § 493.1289 for each specialty and subspecialty of testing performed.
§ 493.1251 Standard: Procedure manual.
(a) A written procedure manual for all tests, assays, and examinations performed by the laboratory must be available to, and followed by, laboratory personnel. Textbooks may supplement but not replace the laboratory's written procedures for testing or examining specimens.
(b) The procedure manual must include the following when applicable to the test procedure:
(1) Requirements for patient preparation; specimen collection, labeling, storage, preservation, transportation, processing, and referral; and criteria for specimen acceptability and rejection as described in § 493.1242.
(2) Microscopic examination, including the detection of inadequately prepared slides.
(3) Step-by-step performance of the procedure, including test calculations and interpretation of results.
(4) Preparation of slides, solutions, calibrators, controls, reagents, stains, and other materials used in testing.
(5) Calibration and calibration verification procedures.
(6) The reportable range for test results for the test system as established or verified in § 493.1253.
(7) Control procedures.
(8) Corrective action to take when calibration or control results fail to meet the laboratory's criteria for acceptability.
(9) Limitations in the test methodology, including interfering substances.
(10) Reference intervals (normal values).
(11) Imminently life-threatening test results, or panic or alert values.
(12) Pertinent literature references.
(13) The laboratory's system for entering results in the patient record and reporting patient results including, when appropriate, the protocol for reporting imminently life-threatening results, or panic, or alert values.
(14) Description of the course of action to take if a test system becomes inoperable.
(c) Manufacturer's test system instructions or operator manuals may be used, when applicable, to meet the requirements of paragraphs (b)(1) through (b)(12) of this section. Any of the items under paragraphs (b)(1) through (b)(12) of this section not provided by the manufacturer must be provided by the laboratory.
(d) Procedures and changes in procedures must be approved, signed, and dated by the current laboratory director before use.
(e) The laboratory must maintain a copy of each procedure with the dates of initial use and discontinuance as described in § 493.1105(a)(2).
[68 FR 3703, Jan. 24, 2003; 68 FR 50724, Aug. 22, 2003]
§ 493.1252 Standard: Test systems, equipment, instruments, reagents, materials, and supplies.
(a) Test systems must be selected by the laboratory. The testing must be performed following the manufacturer's instructions and in a manner that provides test results within the laboratory's stated performance specifications for each test system as determined under § 493.1253.
(b) The laboratory must define criteria for those conditions that are essential for proper storage of reagents and specimens, accurate and reliable test system operation, and test result reporting. The criteria must be consistent with the manufacturer's instructions, if provided. These conditions must be monitored and documented and, if applicable, include the following:
(1) Water quality.
(2) Temperature.
(3) Humidity.
(4) Protection of equipment and instruments from fluctuations and interruptions in electrical current that adversely affect patient test results and test reports.
(c) Reagents, solutions, culture media, control materials, calibration materials, and other supplies, as appropriate, must be labeled to indicate the following:
(1) Identity and when significant, titer, strength or concentration.
(2) Storage requirements.
(3) Preparation and expiration dates.
(4) Other pertinent information required for proper use.
(d) Reagents, solutions, culture media, control materials, calibration materials, and other supplies must not be used when they have exceeded their expiration date, have deteriorated, or are of substandard quality.
(e) Components of reagent kits of different lot numbers must not be interchanged unless otherwise specified by the manufacturer.
§ 493.1253 Standard: Establishment and verification of performance specifications.
(a) Applicability. Laboratories are not required to verify or establish performance specifications for any test system used by the laboratory before April 24, 2003.
(b)
(1) Verification of performance specifications. Each laboratory that introduces an unmodified, FDA-cleared or approved test system must do the following before reporting patient test results:
(i) Demonstrate that it can obtain performance specifications comparable to those established by the manufacturer for the following performance characteristics:
(A) Accuracy.
(B) Precision.
(C) Reportable range of test results for the test system.
(ii) Verify that the manufacturer's reference intervals (normal values) are appropriate for the laboratory's patient population.
(2) Establishment of performance specifications. Each laboratory that modifies an FDA-cleared or approved test system, or introduces a test system not subject to FDA clearance or approval (including methods developed in-house and standardized methods such as text book procedures), or uses a test system in which performance specifications are not provided by the manufacturer must, before reporting patient test results, establish for each test system the performance specifications for the following performance characteristics, as applicable:
(i) Accuracy.
(ii) Precision.
(iii) Analytical sensitivity.
(iv) Analytical specificity to include interfering substances.
(v) Reportable range of test results for the test system.
(vi) Reference intervals (normal values).
(vii) Any other performance characteristic required for test performance.
(3) Determination of calibration and control procedures. The laboratory must determine the test system's calibration procedures and control procedures based upon the performance specifications verified or established under paragraph (b)(1) or (b)(2) of this section.
(c) Documentation. The laboratory must document all activities specified in this section.
[68 FR 3703, Jan. 24, 2003; 68 FR 50724, Aug. 22, 2003]
§ 493.1254 Standard: Maintenance and function checks.
(a) Unmodified manufacturer's equipment, instruments, or test systems. The laboratory must perform and document the following:
(1) Maintenance as defined by the manufacturer and with at least the frequency specified by the manufacturer.
(2) Function checks as defined by the manufacturer and with at least the frequency specified by the manufacturer. Function checks must be within the manufacturer's established limits before patient testing is conducted.
(b) Equipment, instruments, or test systems developed in-house, commercially available and modified by the laboratory, or maintenance and function check protocols are not provided by the manufacturer. The laboratory must do the following:
(1)
(i) Establish a maintenance protocol that ensures equipment, instrument, and test system performance that is necessary for accurate and reliable test results and test result reporting.
(ii) Perform and document the maintenance activities specified in paragraph (b)(1)(i) of this section.
(2)
(i) Define a function check protocol that ensures equipment, instrument, and test system performance that is necessary for accurate and reliable test results and test result reporting.
(ii) Perform and document the function checks, including background or baseline checks, specified in paragraph (b)(2)(i) of this section. Function checks must be within the laboratory's established limits before patient testing is conducted.
§ 493.1255 Standard: Calibration and calibration verification procedures.
Calibration and calibration verification procedures are required to substantiate the continued accuracy of the test system throughout the laboratory's reportable range of test results for the test system. Unless otherwise specified in this subpart, for each applicable test system the laboratory must do the following:
(a) Perform and document calibration procedures—
(1) Following the manufacturer's test system instructions, using calibration materials provided or specified, and with at least the frequency recommended by the manufacturer;
(2) Using the criteria verified or established by the laboratory as specified in § 493.1253(b)(3)—
(i) Using calibration materials appropriate for the test system and, if possible, traceable to a reference method or reference material of known value; and
(ii) Including the number, type, and concentration of calibration materials, as well as acceptable limits for and the frequency of calibration; and
(3) Whenever calibration verification fails to meet the laboratory's acceptable limits for calibration verification.
(b) Perform and document calibration verification procedures—
(1) Following the manufacturer's calibration verification instructions;
(2) Using the criteria verified or established by the laboratory under § 493.1253(b)(3)—
(i) Including the number, type, and concentration of the materials, as well as acceptable limits for calibration verification; and
(ii) Including at least a minimal (or zero) value, a mid-point value, and a maximum value near the upper limit of the range to verify the laboratory's reportable range of test results for the test system; and
(3) At least once every 6 months and whenever any of the following occur:
(i) A complete change of reagents for a procedure is introduced, unless the laboratory can demonstrate that changing reagent lot numbers does not affect the range used to report patient test results, and control values are not adversely affected by reagent lot number changes.
(ii) There is major preventive maintenance or replacement of critical parts that may influence test performance.
(iii) Control materials reflect an unusual trend or shift, or are outside of the laboratory's acceptable limits, and other means of assessing and correcting unacceptable control values fail to identify and correct the problem.
(iv) The laboratory's established schedule for verifying the reportable range for patient test results requires more frequent calibration verification.
§ 493.1256 Standard: Control procedures.
(a) For each test system, the laboratory is responsible for having control procedures that monitor the accuracy and precision of the complete analytic process.
(b) The laboratory must establish the number, type, and frequency of testing control materials using, if applicable, the performance specifications verified or established by the laboratory as specified in § 493.1253(b)(3).
(c) The control procedures must—
(1) Detect immediate errors that occur due to test system failure, adverse environmental conditions, and operator performance.
(2) Monitor over time the accuracy and precision of test performance that may be influenced by changes in test system performance and environmental conditions, and variance in operator performance.
(d) Unless CMS approves a procedure, specified in Appendix C of the State Operations Manual (CMS Pub. 7), that provides equivalent quality testing, the laboratory must—
(1) Perform control procedures as defined in this section unless otherwise specified in the additional specialty and subspecialty requirements at §§ 493.1261 through 493.1278.
(2) For each test system, perform control procedures using the number and frequency specified by the manufacturer or established by the laboratory when they meet or exceed the requirements in paragraph (d)(3) of this section.
(3) At least once each day patient specimens are assayed or examined perform the following for—
(i) Each quantitative procedure, include two control materials of different concentrations;
(ii) Each qualitative procedure, include a negative and positive control material;
(iii) Test procedures producing graded or titered results, include a negative control material and a control material with graded or titered reactivity, respectively;
(iv) Each test system that has an extraction phase, include two control materials, including one that is capable of detecting errors in the extraction process; and
(v) Each molecular amplification procedure, include two control materials and, if reaction inhibition is a significant source of false negative results, a control material capable of detecting the inhibition.
(4) For thin layer chromatography—
(i) Spot each plate or card, as applicable, with a calibrator containing all known substances or drug groups, as appropriate, which are identified by thin layer chromatography and reported by the laboratory; and
(ii) Include at least one control material on each plate or card, as applicable, which must be processed through each step of patient testing, including extraction processes.
(5) For each electrophoretic procedure include, concurrent with patient specimens, at least one control material containing the substances being identified or measured.
(6) Perform control material testing as specified in this paragraph before resuming patient testing when a complete change of reagents is introduced; major preventive maintenance is performed; or any critical part that may influence test performance is replaced.
(7) Over time, rotate control material testing among all operators who perform the test.
(8) Test control materials in the same manner as patient specimens.
(9) When using calibration material as a control material, use calibration material from a different lot number than that used to establish a cut-off value or to calibrate the test system.
(10) Establish or verify the criteria for acceptability of all control materials.
(i) When control materials providing quantitative results are used, statistical parameters (for example, mean and standard deviation) for each batch and lot number of control materials must be defined and available.
(ii) The laboratory may use the stated value of a commercially assayed control material provided the stated value is for the methodology and instrumentation employed by the laboratory and is verified by the laboratory.
(iii) Statistical parameters for unassayed control materials must be established over time by the laboratory through concurrent testing of control materials having previously determined statistical parameters.
(e) For reagent, media, and supply checks, the laboratory must do the following:
(1) Check each batch (prepared in-house), lot number (commercially prepared) and shipment of reagents, disks, stains, antisera, (except those specifically referenced in § 493.1261(a)(3)) and identification systems (systems using two or more substrates or two or more reagents, or a combination) when prepared or opened for positive and negative reactivity, as well as graded reactivity, if applicable.
(2) Each day of use (unless otherwise specified in this subpart), test staining materials for intended reactivity to ensure predictable staining characteristics. Control materials for both positive and negative reactivity must be included, as appropriate.
(3) Check fluorescent and immunohistochemical stains for positive and negative reactivity each time of use.
(4) Before, or concurrent with the initial use—
(i) Check each batch of media for sterility if sterility is required for testing;
(ii) Check each batch of media for its ability to support growth and, as appropriate, select or inhibit specific organisms or produce a biochemical response; and
(iii) Document the physical characteristics of the media when compromised and report any deterioration in the media to the manufacturer.
(5) Follow the manufacturer's specifications for using reagents, media, and supplies and be responsible for results.
(f) Results of control materials must meet the laboratory's and, as applicable, the manufacturer's test system criteria for acceptability before reporting patient test results.
(g) The laboratory must document all control procedures performed.
(h) If control materials are not available, the laboratory must have an alternative mechanism to detect immediate errors and monitor test system performance over time. The performance of alternative control procedures must be documented.
[68 FR 3703, Jan. 24, 2003; 68 FR 50724, Aug. 22, 2003]
§ 493.1261 Standard: Bacteriology.
(a) The laboratory must check the following for positive and negative reactivity using control organisms:
(1) Each day of use for beta-lactamase methods other than CefinaseTM.
(2) Each week of use for Gram stains.
(3) When each batch (prepared in-house), lot number (commercially prepared), and shipment of antisera is prepared or opened, and once every 6 months thereafter.
(b) For antimicrobial susceptibility tests, the laboratory must check each batch of media and each lot number and shipment of antimicrobial agent(s) before, or concurrent with, initial use, using approved control organisms.
(1) Each day tests are performed, the laboratory must use the appropriate control organism(s) to check the procedure.
(2) The laboratory's zone sizes or minimum inhibitory concentration for control organisms must be within established limits before reporting patient results.
(c) The laboratory must document all control procedures performed, as specified in this section.
§ 493.1262 Standard: Mycobacteriology.
(a) Each day of use, the laboratory must check all reagents or test procedures used for mycobacteria identification with at least one acid-fast organism that produces a positive reaction and an acid-fast organism that produces a negative reaction.
(b) For antimycobacterial susceptibility tests, the laboratory must check each batch of media and each lot number and shipment of antimycobacterial agent(s) before, or concurrent with, initial use, using an appropriate control organism(s).
(1) The laboratory must establish limits for acceptable control results.
(2) Each week tests are performed, the laboratory must use the appropriate control organism(s) to check the procedure.
(3) The results for the control organism(s) must be within established limits before reporting patient results.
(c) The laboratory must document all control procedures performed, as specified in this section.
§ 493.1263 Standard: Mycology.
(a) The laboratory must check each batch (prepared in-house), lot number (commercially prepared), and shipment of lactophenol cotton blue when prepared or opened for intended reactivity with a control organism(s).
(b) For antifungal susceptibility tests, the laboratory must check each batch of media and each lot number and shipment of antifungal agent(s) before, or concurrent with, initial use, using an appropriate control organism(s).
(1) The laboratory must establish limits for acceptable control results.
(2) Each day tests are performed, the laboratory must use the appropriate control organism(s) to check the procedure.
(3) The results for the control organism(s) must be within established limits before reporting patient results.
(c) The laboratory must document all control procedures performed, as specified in this section.
§ 493.1264 Standard: Parasitology.
(a) The laboratory must have available a reference collection of slides or photographs and, if available, gross specimens for identification of parasites and use these references in the laboratory for appropriate comparison with diagnostic specimens.
(b) The laboratory must calibrate and use the calibrated ocular micrometer for determining the size of ova and parasites, if size is a critical parameter.
(c) Each month of use, the laboratory must check permanent stains using a fecal sample control material that will demonstrate staining characteristics.
(d) The laboratory must document all control procedures performed, as specified in this section.
§ 493.1265 Standard: Virology.
(a) When using cell culture to isolate or identify viruses, the laboratory must simultaneously incubate a cell substrate control or uninoculated cells as a negative control material.
(b) The laboratory must document all control procedures performed, as specified in this section.
§ 493.1267 Standard: Routine chemistry.
For blood gas analyses, the laboratory must perform the following:
(a) Calibrate or verify calibration according to the manufacturer's specifications and with at least the frequency recommended by the manufacturer.
(b) Test one sample of control material each 8 hours of testing using a combination of control materials that include both low and high values on each day of testing.
(c) Test one sample of control material each time specimens are tested unless automated instrumentation internally verifies calibration at least every 30 minutes.
(d) Document all control procedures performed, as specified in this section.
§ 493.1269 Standard: Hematology.
(a) For manual cell counts performed using a hemocytometer—
(1) One control material must be tested each 8 hours of operation; and
(2) Patient specimens and control materials must be tested in duplicate.
(b) For all nonmanual coagulation test systems, the laboratory must include two levels of control material each 8 hours of operation and each time a reagent is changed.
(c) For manual coagulation tests—
(1) Each individual performing tests must test two levels of control materials before testing patient samples and each time a reagent is changed; and
(2) Patient specimens and control materials must be tested in duplicate.
(d) The laboratory must document all control procedures performed, as specified in this section.
§ 493.1271 Standard: Immunohematology.
(a) Patient testing.
(1) The laboratory must perform ABO grouping, D(Rho) typing, unexpected antibody detection, antibody identification, and compatibility testing by following the manufacturer's instructions, if provided, and as applicable, 21 CFR 606.151(a) through (e).
(2) The laboratory must determine ABO group by concurrently testing unknown red cells with, at a minimum, anti-A and anti-B grouping reagents. For confirmation of ABO group, the unknown serum must be tested with known A1 and B red cells.
(3) The laboratory must determine the D(Rho) type by testing unknown red cells with anti-D (anti-Rho) blood typing reagent.
(b) Immunohematological testing and distribution of blood and blood products. Blood and blood product testing and distribution must comply with 21 CFR 606.100(b)(12); 606.160(b)(3)(ii) and (b)(3)(v); 610.40; 640.5(a), (b), (c), and (e); and 640.11(b).
(c) Blood and blood products storage. Blood and blood products must be stored under appropriate conditions that include an adequate temperature alarm system that is regularly inspected.
(1) An audible alarm system must monitor proper blood and blood product storage temperature over a 24-hour period.
(2) Inspections of the alarm system must be documented.
(d) Retention of samples of transfused blood. According to the laboratory's established procedures, samples of each unit of transfused blood must be retained for further testing in the event of transfusion reactions. The laboratory must promptly dispose of blood not retained for further testing that has passed its expiration date.
(e) Investigation of transfusion reactions.
(1) According to its established procedures, the laboratory that performs compatibility testing, or issues blood or blood products, must promptly investigate all transfusion reactions occurring in facilities for which it has investigational responsibility and make recommendations to the medical staff regarding improvements in transfusion procedures.
(2) The laboratory must document, as applicable, that all necessary remedial actions are taken to prevent recurrences of transfusion reactions and that all policies and procedures are reviewed to assure they are adequate to ensure the safety of individuals being transfused.
(f) Documentation. The laboratory must document all control procedures performed, as specified in this section.
[68 FR 3703, Jan. 24, 2003; 68 FR 50724, Aug. 22, 2003]
§ 493.1273 Standard: Histopathology.
(a) As specified in § 493.1256(e)(3), fluorescent and immunohistochemical stains must be checked for positive and negative reactivity each time of use. For all other differential or special stains, a control slide of known reactivity must be stained with each patient slide or group of patient slides. Reaction(s) of the control slide with each special stain must be documented.
(b) The laboratory must retain stained slides, specimen blocks, and tissue remnants as specified in § 493.1105. The remnants of tissue specimens must be maintained in a manner that ensures proper preservation of the tissue specimens until the portions submitted for microscopic examination have been examined and a diagnosis made by an individual qualified under § 493.1449(b), (l), or (m).
(c) An individual who has successfully completed a training program in neuromuscular pathology approved by HHS may examine and provide reports for neuromuscular pathology.
(d) Tissue pathology reports must be signed by an individual qualified as specified in paragraph (b) or, as appropriate, paragraph (c) of this section. If a computer report is generated with an electronic signature, it must be authorized by the individual who performed the examination and made the diagnosis.
(e) The laboratory must use acceptable terminology of a recognized system of disease nomenclature in reporting results.
(f) The laboratory must document all control procedures performed, as specified in this section.
[68 FR 3703, Jan. 24, 2003; 68 FR 50724, Aug. 22, 2003]
§ 493.1274 Standard: Cytology.
(a) Cytology slide examination site. All cytology slide preparations must be evaluated on the premises of a laboratory certified to conduct testing in the subspecialty of cytology.
(b) Staining. The laboratory must have available and follow written policies and procedures for each of the following, if applicable:
(1) All gynecologic slide preparations must be stained using a Papanicolaou or modified Papanicolaou staining method.
(2) Effective measures to prevent cross-contamination between gynecologic and nongynecologic specimens during the staining process must be used.
(3) Nongynecologic specimens that have a high potential for cross-contamination must be stained separately from other nongynecologic specimens, and the stains must be filtered or changed following staining.
(c) Control procedures. The laboratory must establish and follow written policies and procedures for a program designed to detect errors in the performance of cytologic examinations and the reporting of results. The program must include the following:
(1) A review of slides from at least 10 percent of the gynecologic cases interpreted by individuals qualified under § 493.1469 or § 493.1483, to be negative for epithelial cell abnormalities and other malignant neoplasms (as defined in paragraph (e)(1) of this section).
(i) The review must be performed by an individual who meets one of the following qualifications:
(A) A technical supervisor qualified under § 493.1449(b) or (k).
(B) A cytology general supervisor qualified under § 493.1469.
(C) A cytotechnologist qualified under § 493.1483 who has the experience specified in § 493.1469(b)(2).
(ii) Cases must be randomly selected from the total caseload and include negatives and those from patients or groups of patients that are identified as having a higher than average probability of developing cervical cancer based on available patient information.
(iii) The review of those cases selected must be completed before reporting patient results.
(2) Laboratory comparison of clinical information, when available, with cytology reports and comparison of all gynecologic cytology reports with a diagnosis of high-grade squamous intraepithelial lesion (HSIL), adenocarcinoma, or other malignant neoplasms with the histopathology report, if available in the laboratory (either on-site or in storage), and determination of the causes of any discrepancies.
(3) For each patient with a current HSIL, adenocarcinoma, or other malignant neoplasm, laboratory review of all normal or negative gynecologic specimens received within the previous 5 years, if available in the laboratory (either on-site or in storage). If significant discrepancies are found that will affect current patient care, the laboratory must notify the patient's physician and issue an amended report.
(4) Records of initial examinations and all rescreening results must be documented.
(5) An annual statistical laboratory evaluation of the number of—
(i) Cytology cases examined;
(ii) Specimens processed by specimen type;
(iii) Patient cases reported by diagnosis (including the number reported as unsatisfactory for diagnostic interpretation);
(iv) Gynecologic cases with a diagnosis of HSIL, adenocarcinoma, or other malignant neoplasm for which histology results were available for comparison;
(v) Gynecologic cases where cytology and histology are discrepant; and
(vi) Gynecologic cases where any rescreen of a normal or negative specimen results in reclassification as low-grade squamous intraepithelial lesion (LSIL), HSIL, adenocarcinoma, or other malignant neoplasms.
(6) An evaluation of the case reviews of each individual examining slides against the laboratory's overall statistical values, documentation of any discrepancies, including reasons for the deviation and, if appropriate, corrective actions taken.
(d) Workload limits. The laboratory must establish and follow written policies and procedures that ensure the following:
(1) The technical supervisor establishes a maximum workload limit for each individual who performs primary screening.
(i) The workload limit is based on the individual's performance using evaluations of the following:
(A) Review of 10 percent of the cases interpreted as negative for the conditions defined in paragraph (e)(1) of this section.
(B) Comparison of the individual's interpretation with the technical supervisor's confirmation of patient smears specified in paragraphs (e)(1) and (e)(3) of this section.
(ii) Each individual's workload limit is reassessed at least every 6 months and adjusted when necessary.
(2) The maximum number of slides examined by an individual in each 24-hour period does not exceed 100 slides (one patient specimen per slide; gynecologic, nongynecologic, or both) irrespective of the site or laboratory. This limit represents an absolute maximum number of slides and must not be employed as an individual's performance target. In addition—
(i) The maximum number of 100 slides is examined in no less than an 8-hour workday;
(ii) For the purposes of establishing workload limits for individuals examining slides in less than an 8-hour workday (includes full-time employees with duties other than slide examination and part-time employees), a period of 8 hours is used to prorate the number of slides that may be examined. The formula—
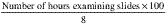
is used to determine maximum slide volume to be examined;
(iii) Nongynecologic slide preparations made using liquid-based slide preparatory techniques that result in cell dispersion over one-half or less of the total available slide may be counted as one-half slide; and
(iv) Technical supervisors who perform primary screening are not required to include tissue pathology slides and previously examined cytology slides (gynecologic and nongynecologic) in the 100 slide workload limit.
(3) The laboratory must maintain records of the total number of slides examined by each individual during each 24-hour period and the number of hours spent examining slides in the 24-hour period irrespective of the site or laboratory.
(4) Records are available to document the workload limit for each individual.
(e) Slide examination and reporting. The laboratory must establish and follow written policies and procedures that ensure the following:
(1) A technical supervisor confirms each gynecologic slide preparation interpreted to exhibit reactive or reparative changes or any of the following epithelial cell abnormalities:
(i) Squamous cell.
(A) Atypical squamous cells of undetermined significance (ASC-US) or cannot exclude HSIL (ASC-H).
(B) LSIL-Human papillomavirus (HPV)/mild dysplasia/cervical intraepithelial neoplasia 1 (CIN 1).
(C) HSIL-moderate and severe dysplasia, carcinoma in situ (CIS)/CIN 2 and CIN 3 or with features suspicious for invasion.
(D) Squamous cell carcinoma.
(ii) Glandular cell.
(A) Atypical cells not otherwise specified (NOS) or specified in comments (endocervical, endometrial, or glandular).
(B) Atypical cells favor neoplastic (endocervical or glandular).
(C) Endocervical adenocarcinoma in situ.
(D) Adenocarcinoma endocervical, adenocarcinoma endometrial, adenocarcinoma extrauterine, and adenocarcinoma NOS.
(iii) Other malignant neoplasms.
(2) The report of gynecologic slide preparations with conditions specified in paragraph (e)(1) of this section must be signed to reflect the technical supervisory review or, if a computer report is generated with signature, it must reflect an electronic signature authorized by the technical supervisor who performed the review.
(3) All nongynecologic preparations are reviewed by a technical supervisor. The report must be signed to reflect technical supervisory review or, if a computer report is generated with signature, it must reflect an electronic signature authorized by the technical supervisor who performed the review.
(4) Unsatisfactory specimens or slide preparations are identified and reported as unsatisfactory.
(5) The report contains narrative descriptive nomenclature for all results.
(6) Corrected reports issued by the laboratory indicate the basis for correction.
(f) Record and slide retention.
(1) The laboratory must retain all records and slide preparations as specified in § 493.1105.
(2) Slides may be loaned to proficiency testing programs in lieu of maintaining them for the required time period, provided the laboratory receives written acknowledgment of the receipt of slides by the proficiency testing program and maintains the acknowledgment to document the loan of these slides.
(3) Documentation of slides loaned or referred for purposes other than proficiency testing must be maintained.
(4) All slides must be retrievable upon request.
(g) Automated and semi-automated screening devices. When performing evaluations using automated and semi-automated screening devices, the laboratory must follow manufacturer's instructions for preanalytic, analytic, and postanalytic phases of testing, as applicable, and meet the applicable requirements of this subpart K.
(h) Documentation. The laboratory must document all control procedures performed, as specified in this section.
[68 FR 3703, Jan. 24, 2003; 68 FR 50724, Aug. 22, 2003]
§ 493.1276 Standard: Clinical cytogenetics.
(a) The laboratory must have policies and procedures for ensuring accurate and reliable patient specimen identification during the process of accessioning, cell preparation, photographing or other image reproduction technique, photographic printing, and reporting and storage of results, karyotypes, and photographs.
(b) The laboratory must have records that document the following:
(1) The media used, reactions observed, number of cells counted, number of cells karyotyped, number of chromosomes counted for each metaphase spread, and the quality of the banding.
(2) The resolution is appropriate for the type of tissue or specimen and the type of study required based on the clinical information provided to the laboratory.
(3) An adequate number of karyotypes are prepared for each patient.
(c) Determination of sex must be performed by full chromosome analysis.
(d) The laboratory report must include a summary and interpretation of the observations, number of cells counted and analyzed, and use the International System for Human Cytogenetic Nomenclature.
(e) The laboratory must document all control procedures performed, as specified in this section.
[68 FR 3703, Jan. 24, 2003; 68 FR 50724, Aug. 22, 2003]
§ 493.1278 Standard: Histocompatibility.
(a) General. The laboratory must meet the following requirements:
(1) An audible alarm system must be used to monitor the storage temperature of specimens (donor and beneficiary) and reagents. The laboratory must have an emergency plan for alternate storage.
(2) All patient specimens must be easily retrievable.
(3) Reagent typing sera inventory prepared in-house must indicate source, bleeding date and identification number, reagent specificity, and volume remaining.
(4) If the laboratory uses immunologic reagents (for example, antibodies, antibody-coated particles, or complement) to facilitate or enhance the isolation of lymphocytes, or lymphocyte subsets, the efficacy of the methods must be monitored with appropriate quality control procedures.
(5) Participate in at least one national or regional cell exchange program, if available, or develop an exchange system with another laboratory in order to validate interlaboratory reproducibility.
(b) HLA typing. The laboratory must do the following:
(1) Use a technique(s) that is established to optimally define, as applicable, HLA Class I and II specificities.
(2) HLA type all potential transplant beneficiaries at a level appropriate to support clinical transplant protocol and donor selection.
(3) HLA type cells from organ donors referred to the laboratory.
(4) Use HLA antigen terminology that conforms to the latest report of the World Health Organization (W.H.O.) Committee on Nomenclature. Potential new antigens not yet approved by this committee must have a designation that cannot be confused with W.H.O. terminology.
(5) Have available and follow written criteria for the following:
(i) The preparation of cells or cellular extracts (for example, solubilized antigens and nucleic acids), as applicable to the HLA typing technique(s) performed.
(ii) Selecting typing reagents, whether prepared in-house or commercially.
(iii) Ensuring that reagents used for typing are adequate to define all HLA-A, B and DR specificities that are officially recognized by the most recent W.H.O. Committee on Nomenclature and for which reagents are readily available.
(iv) The assignment of HLA antigens.
(v) When antigen redefinition and retyping are required.
(6) Check each HLA typing by testing, at a minimum the following:
(i) A positive control material.
(ii) A negative control material in which, if applicable to the technique performed, cell viability at the end of incubation is sufficient to permit accurate interpretation of results. In assays in which cell viability is not required, the negative control result must be sufficiently different from the positive control result to permit accurate interpretation of results.
(iii) Positive control materials for specific cell types when applicable (that is, T cells, B cells, and monocytes).
(c) Disease-associated studies. The laboratory must check each typing for disease-associated HLA antigens using control materials to monitor the test components and each phase of the test system to ensure acceptable performance.
(d) Antibody Screening. The laboratory must do the following:
(1) Use a technique(s) that detects HLA-specific antibody with a specificity equivalent or superior to that of the basic complement-dependent microlymphocytotoxicity assay.
(2) Use a method that distinguishes antibodies to HLA Class II antigens from antibodies to Class I antigens to detect antibodies to HLA Class II antigens.
(3) Use a panel that contains all the major HLA specificities and common splits. If the laboratory does not use commercial panels, it must maintain a list of individuals for fresh panel bleeding.
(4) Make a reasonable attempt to have available monthly serum specimens for all potential transplant beneficiaries for periodic antibody screening and crossmatch.
(5) Have available and follow a written policy consistent with clinical transplant protocols for the frequency of screening potential transplant beneficiary sera for preformed HLA-specific antibodies.
(6) Check each antibody screening by testing, at a minimum the following:
(i) A positive control material containing antibodies of the appropriate isotype for the assay.
(ii) A negative control material.
(7) As applicable, have available and follow written criteria and procedures for antibody identification to the level appropriate to support clinical transplant protocol.
(e) Crossmatching. The laboratory must do the following:
(1) Use a technique(s) documented to have increased sensitivity in comparison with the basic complement-dependent microlymphocytotoxicity assay.
(2) Have available and follow written criteria for the following:
(i) Selecting appropriate patient serum samples for crossmatching.
(ii) The preparation of donor cells or cellular extracts (for example, solubilized antigens and nucleic acids), as applicable to the crossmatch technique(s) performed.
(3) Check each crossmatch and compatibility test for HLA Class II antigenic differences using control materials to monitor the test components and each phase of the test system to ensure acceptable performance.
(f) Transplantation. Laboratories performing histocompatibility testing for transfusion and transplantation purposes must do the following:
(1) Have available and follow written policies and protocols specifying the histocompatibility testing (that is, HLA typing, antibody screening, compatibility testing and crossmatching) to be performed for each type of cell, tissue or organ to be transfused or transplanted. The laboratory's policies must include, as applicable—
(i) Testing protocols for cadaver donor, living, living-related, and combined organ and tissue transplants;
(ii) Testing protocols for patients at high risk for allograft rejection; and
(iii) The level of testing required to support clinical transplant protocols (for example, antigen or allele level).
(2) For renal allotransplantation and combined organ and tissue transplants in which a kidney is to be transplanted, have available results of final crossmatches before the kidney is transplanted.
(3) For nonrenal transplantation, if HLA testing and final crossmatches were not performed prospectively because of an emergency situation, the laboratory must document the circumstances, if known, under which the emergency transplant was performed, and records of the transplant must reflect any information provided to the laboratory by the patient's physician.
(g) Documentation. The laboratory must document all control procedures performed, as specified in this section.
[68 FR 3703, Jan. 24, 2003; 68 FR 50724, Aug. 22, 2003]
§ 493.1281 Standard: Comparison of test results.
(a) If a laboratory performs the same test using different methodologies or instruments, or performs the same test at multiple testing sites, the laboratory must have a system that twice a year evaluates and defines the relationship between test results using the different methodologies, instruments, or testing sites.
(b) The laboratory must have a system to identify and assess patient test results that appear inconsistent with the following relevant criteria, when available:
(1) Patient age.
(2) Sex.
(3) Diagnosis or pertinent clinical data.
(4) Distribution of patient test results.
(5) Relationship with other test parameters.
(c) The laboratory must document all test result comparison activities.
§ 493.1282 Standard: Corrective actions.
(a) Corrective action policies and procedures must be available and followed as necessary to maintain the laboratory's operation for testing patient specimens in a manner that ensures accurate and reliable patient test results and reports.
(b) The laboratory must document all corrective actions taken, including actions taken when any of the following occur:
(1) Test systems do not meet the laboratory's verified or established performance specifications, as determined in § 493.1253(b), which include but are not limited to—
(i) Equipment or methodologies that perform outside of established operating parameters or performance specifications;
(ii) Patient test values that are outside of the laboratory's reportable range of test results for the test system; and
(iii) When the laboratory determines that the reference intervals (normal values) for a test procedure are inappropriate for the laboratory's patient population.
(2) Results of control or calibration materials, or both, fail to meet the laboratory's established criteria for acceptability. All patient test results obtained in the unacceptable test run and since the last acceptable test run must be evaluated to determine if patient test results have been adversely affected. The laboratory must take the corrective action necessary to ensure the reporting of accurate and reliable patient test results.
(3) The criteria for proper storage of reagents and specimens, as specified under § 493.1252(b), are not met.
§ 493.1283 Standard: Test records.
(a) The laboratory must maintain an information or record system that includes the following:
(1) The positive identification of the specimen.
(2) The date and time of specimen receipt into the laboratory.
(3) The condition and disposition of specimens that do not meet the laboratory's criteria for specimen acceptability.
(4) The records and dates of all specimen testing, including the identity of the personnel who performed the test(s).
(b) Records of patient testing including, if applicable, instrument printouts, must be retained.
§ 493.1289 Standard: Analytic systems quality assessment.
(a) The laboratory must establish and follow written policies and procedures for an ongoing mechanism to monitor, assess, and when indicated, correct problems identified in the analytic systems specified in §§ 493.1251 through 493.1283.
(b) The analytic systems quality assessment must include a review of the effectiveness of corrective actions taken to resolve problems, revision of policies and procedures necessary to prevent recurrence of problems, and discussion of analytic systems quality assessment reviews with appropriate staff.
(c) The laboratory must document all analytic systems quality assessment activities.
[68 FR 3703, Jan. 24, 2003; 68 FR 50724, Aug. 22, 2003]
Postanalytic Systems
§ 493.1290 Condition: Postanalytic systems.
Each laboratory that performs nonwaived testing must meet the applicable postanalytic systems requirements in § 493.1291 unless HHS approves a procedure, specified in Appendix C of the State Operations Manual (CMS Pub. 7) that provides equivalent quality testing. The laboratory must monitor and evaluate the overall quality of the postanalytic systems and correct identified problems as specified in § 493.1299 for each specialty and subspecialty of testing performed.
§ 493.1291 Standard: Test report.
(a) The laboratory must have an adequate manual or electronic system(s) in place to ensure test results and other patient-specific data are accurately and reliably sent from the point of data entry (whether interfaced or entered manually) to final report destination, in a timely manner. This includes the following:
(1) Results reported from calculated data.
(2) Results and patient-specific data electronically reported to network or interfaced systems.
(3) Manually transcribed or electronically transmitted results and patient-specific information reported directly or upon receipt from outside referral laboratories, satellite or point-of-care testing locations.
(b) Test report information maintained as part of the patient's chart or medical record must be readily available to the laboratory and to CMS or a CMS agent upon request.
(c) The test report must indicate the following:
(1) For positive patient identification, either the patient's name and identification number, or a unique patient identifier and identification number.
(2) The name and address of the laboratory location where the test was performed.
(3) The test report date.
(4) The test performed.
(5) Specimen source, when appropriate.
(6) The test result and, if applicable, the units of measurement or interpretation, or both.
(7) Any information regarding the condition and disposition of specimens that do not meet the laboratory's criteria for acceptability.
(d) Pertinent “reference intervals” or “normal” values, as determined by the laboratory performing the tests, must be available to the authorized person who ordered the tests and, if applicable, the individual responsible for using the test results.
(e) The laboratory must, upon request, make available to clients a list of test methods employed by the laboratory and, as applicable, the performance specifications established or verified as specified in § 493.1253. In addition, information that may affect the interpretation of test results, for example test interferences, must be provided upon request. Pertinent updates on testing information must be provided to clients whenever changes occur that affect the test results or interpretation of test results.
(f) Except as provided in § 493.1291(l), test results must be released only to authorized persons and, if applicable, the persons responsible for using the test results and the laboratory that initially requested the test.
(g) The laboratory must immediately alert the individual or entity requesting the test and, if applicable, the individual responsible for using the test results when any test result indicates an imminently life-threatening condition, or panic or alert values.
(h) When the laboratory cannot report patient test results within its established time frames, the laboratory must determine, based on the urgency of the patient test(s) requested, the need to notify the appropriate individual(s) of the delayed testing.
(i) If a laboratory refers patient specimens for testing—
(1) The referring laboratory must not revise results or information directly related to the interpretation of results provided by the testing laboratory;
(2) The referring laboratory may permit each testing laboratory to send the test result directly to the authorized person who initially requested the test. The referring laboratory must retain or be able to produce an exact duplicate of each testing laboratory's report; and
(3) The authorized person who orders a test must be notified by the referring laboratory of the name and address of each laboratory location where the test was performed.
(j) All test reports or records of the information on the test reports must be maintained by the laboratory in a manner that permits ready identification and timely accessibility.
(k) When errors in the reported patient test results are detected, the laboratory must do the following:
(1) Promptly notify the authorized person ordering the test and, if applicable, the individual using the test results of reporting errors.
(2) Issue corrected reports promptly to the authorized person ordering the test and, if applicable, the individual using the test results.
(3) Maintain duplicates of the original report, as well as the corrected report.
(l) Upon request by a patient (or the patient's personal representative), the laboratory may provide patients, their personal representatives, and those persons specified under 45 CFR 164.524(c)(3)(ii), as applicable, with access to completed test reports that, using the laboratory's authentication process, can be identified as belonging to that patient.
[68 FR 3703, Jan. 24, 2003; 68 FR 50724, Aug. 22, 2003, as amended at 79 FR 7316, Feb. 6, 2014]
§ 493.1299 Standard: Postanalytic systems quality assessment.
(a) The laboratory must establish and follow written policies and procedures for an ongoing mechanism to monitor, assess and, when indicated, correct problems identified in the postanalytic systems specified in § 493.1291.
(b) The postanalytic systems quality assessment must include a review of the effectiveness of corrective actions taken to resolve problems, revision of policies and procedures necessary to prevent recurrence of problems, and discussion of postanalytic systems quality assessment reviews with appropriate staff.
(c) The laboratory must document all postanalytic systems quality assessment activities.
[68 FR 3703, Jan. 24, 2003; 68 FR 50724, Aug. 22, 2003]
Subpart L [Reserved]
Subpart M - Personnel for Nonwaived Testing
Source:
57 FR 7172, Feb. 28, 1992, unless otherwise noted.
§ 493.1351 General.
This subpart consists of the personnel requirements that must be met by laboratories performing moderate complexity testing, PPM procedures, high complexity testing, or any combination of these tests.
[60 FR 20049, Apr. 24, 1995]
Laboratories Performing Provider-Performed Microscopy (PPM) Procedures
Source:
60 FR 20049, Apr. 24, 1995, unless otherwise noted.
§ 493.1353 Scope.
In accordance with § 493.19(b), the moderate complexity procedures specified as PPM procedures are considered such only when personally performed by a health care provider during a patient visit in the context of a physical examination. PPM procedures are subject to the personnel requirements in §§ 493.1355 through 493.1365.
§ 493.1355 Condition: Laboratories performing PPM procedures; laboratory director.
The laboratory must have a director who meets the qualification requirements of § 493.1357 and provides overall management and direction in accordance with § 493.1359.
§ 493.1357 Standard; laboratory director qualifications.
The laboratory director must be qualified to manage and direct the laboratory personnel and the performance of PPM procedures as specified in § 493.19(c) and must be eligible to be an operator of a laboratory within the requirements of subpart R of this part.
(a) The laboratory director must possess a current license as a laboratory director issued by the State in which the laboratory is located, if the licensing is required.
(b) The laboratory director must meet one of the following requirements:
(1) Be a physician, as defined in § 493.2.
(2) Be a midlevel practitioner, as defined in § 493.2, authorized by a State to practice independently in the State in which the laboratory is located.
(3) Be a dentist, as defined in § 493.2.
§ 493.1359 Standard; PPM laboratory director responsibilities.
The laboratory director is responsible for the overall operation and administration of the laboratory, including the prompt, accurate, and proficient reporting of test results. The laboratory director must—
(a) Direct no more than five laboratories; and
(b) Ensure that any procedure listed under § 493.19(c)—
(1) Is personally performed by an individual who meets the qualification requirements in § 493.1363; and
(2) Is performed in accordance with applicable requirements in subparts H, J, K, and M of this part.
[57 FR 7172, Feb. 28, 1992, as amended at 68 FR 3713, Jan. 24, 2003; 68 FR 50724, Aug. 22, 2003]
§ 493.1361 Condition: Laboratories performing PPM procedures; testing personnel.
The laboratory must have a sufficient number of individuals who meet the qualification requirements of § 493.1363 to perform the functions specified in § 493.1365 for the volume and complexity of testing performed.
§ 493.1363 Standard: PPM testing personnel qualifications.
Each individual performing PPM procedures must—
(a) Possess a current license issued by the State in which the laboratory is located if the licensing is required; and
(b) Meet one of the following requirements:
(1) Be a physician, as defined in § 493.2.
(2) Be a midlevel practitioner, as defined in § 493.2, under the supervision of a physician or in independent practice if authorized by the State in which the laboratory is located.
(3) Be a dentist as defined in § 493.2 of this part.
§ 493.1365 Standard; PPM testing personnel responsibilities.
The testing personnel are responsible for specimen processing, test performance, and for reporting test results. Any PPM procedure must be—
(a) Personally performed by one of the following practitioners:
(1) A physician during the patient's visit on a specimen obtained from his or her own patient or from a patient of a group medical practice of which the physician is a member or employee.
(2) A midlevel practitioner, under the supervision of a physician or in independent practice if authorized by the State in which the laboratory is located, during the patient's visit on a specimen obtained from his or her own patient or from the patient of a clinic, group medical practice, or other health care provider, in which the midlevel practitioner is a member or an employee.
(3) A dentist during the patient's visit on a specimen obtained from his or her own patient or from a patient of a group dental practice of which the dentist is a member or an employee; and
(b) Performed using a microscope limited to a brightfield or a phase/contrast microscope.
Laboratories Performing Moderate Complexity Testing
§ 493.1403 Condition: Laboratories performing moderate complexity testing; laboratory director.
The laboratory must have a director who meets the qualification requirements of § 493.1405 of this subpart and provides overall management and direction in accordance with § 493.1407 of this subpart.
§ 493.1405 Standard; Laboratory director qualifications.
The laboratory director must be qualified to manage and direct the laboratory personnel and the performance of moderate complexity tests and must be eligible to be an operator of a laboratory within the requirements of subpart R of this part.
(a) The laboratory director must possess a current license as a laboratory director issued by the State in which the laboratory is located, if such licensing is required; and
(b) The laboratory director must—
(1)
(i) Be a doctor of medicine or doctor of osteopathy licensed to practice medicine or osteopathy in the State in which the laboratory is located; and
(ii) Be certified in anatomic or clinical pathology, or both, by the American Board of Pathology or the American Osteopathic Board of Pathology or possess qualifications that are equivalent to those required for such certification; or
(2)
(i) Be a doctor of medicine, doctor of osteopathy, or doctor of podiatric medicine licensed to practice medicine, osteopathy, or podiatry in the State in which the laboratory is located; and
(ii) Have had laboratory training or experience consisting of:
(A) At least one year directing or supervising non-waived laboratory testing; or
(B) Beginning September 1, 1993, have at least 20 continuing medical education credit hours in laboratory practice commensurate with the director responsibilities defined in § 493.1407; or
(C) Laboratory training equivalent to paragraph (b)(2)(ii)(B) of this section obtained during medical residency. (For example, physicians certified either in hematology or hematology and medical oncology by the American Board of Internal Medicine); or
(3) Hold an earned doctoral degree in a chemical, physical, biological, or clinical laboratory science from an accredited institution; and
(i) Be certified by the American Board of Medical Microbiology, the American Board of Clinical Chemistry, the American Board of Bioanalysis, or the American Board of Medical Laboratory Immunology; or
(ii) Have had at least one year experience directing or supervising non-waived laboratory testing;
(4)
(i) Have earned a master's degree in a chemical, physical, biological or clinical laboratory science or medical technology from an accredited institution;
(ii) Have at least one year of laboratory training or experience, or both in non-waived testing; and
(iii) In addition, have at least one year of supervisory laboratory experience in non-waived testing; or
(5)
(i) Have earned a bachelor's degree in a chemical, physical, or biological science or medical technology from an accredited institution;
(ii) Have at least 2 years of laboratory training or experience, or both in non-waived testing; and
(iii) In addition, have at least 2 years of supervisory laboratory experience in non-waived testing;
(6) Be serving as a laboratory director and must have previously qualified or could have qualified as a laboratory director under § 493.1406; or
(7) On or before February 28, 1992, qualified under State law to direct a laboratory in the State in which the laboratory is located.
[57 FR 7172, Feb. 28, 1992, as amended at 58 FR 5233, Jan. 19, 1993]
§ 493.1406 Standard; Laboratory director qualifications on or before February 28, 1992.
The laboratory director must be qualified to manage and direct the laboratory personnel and test performance.
(a) The laboratory director must possess a current license as a laboratory director issued by the State, if such licensing exists; and
(b) The laboratory director must:
(1) Be a physician certified in anatomical or clinical pathology (or both) by the American Board of Pathology or the American Osteopathic Board of Pathology or possess qualifications that are equivalent to those required for such certification;
(2) Be a physician who:
(i) Is certified by the American Board of Pathology or the American Osteopathic Board of Pathology in at least one of the laboratory specialties; or
(ii) Is certified by the American Board of Medical Microbiology, the American Board of Clinical Chemistry, the American Board of Bioanalysis, or other national accrediting board in one of the laboratory specialties; or
(iii) Is certified by the American Society of Cytology to practice cytopathology or possesses qualifications that are equivalent to those required for such certification; or
(iv) Subsequent to graduation, has had 4 or more years of full-time general laboratory training and experience of which at least 2 years were spent acquiring proficiency in one of the laboratory specialties;
(3) For the subspecialty of oral pathology only, be certified by the American Board of Oral Pathology, American Board of Pathology or the American Osteopathic Board of Pathology or possesses qualifications that are equivalent to those required for certification;
(4) Hold an earned doctoral degree from an accredited institution with a chemical, physical, or biological science as a major subject and
(i) Is certified by the American Board of Medical Microbiology, the American Board of Clinical Chemistry, the American Board of Bioanalysis, or other national accrediting board acceptable to HHS in one of the laboratory specialties; or
(ii) Subsequent to graduation, has had 4 or more years of full-time general laboratory training and experience of which at least 2 years were spent acquiring proficiency in one of the laboratory specialties;
(5) With respect to individuals first qualifying before July 1, 1971, have been responsible for the direction of a laboratory for 12 months between July 1, 1961, and January 1, 1968, and, in addition, either:
(i) Was a physician and subsequent to graduation had at least 4 years of pertinent full-time laboratory experience;
(ii) Held a master's degree from an accredited institution with a chemical, physical, or biological science as a major subject and subsequent to graduation had at least 4 years of pertinent full-time laboratory experience;
(iii) Held a bachelor's degree from an accredited institution with a chemical, physical, or biological science as a major subject and subsequent to graduation had at least 6 years of pertinent full-time laboratory experience; or
(iv) Achieved a satisfactory grade through an examination conducted by or under the sponsorship of the U.S. Public Health Service on or before July 1, 1970; or
(6) Qualify under State law to direct the laboratory in the State in which the laboratory is located.
The January 1, 1968 date for meeting the 12 months' laboratory direction requirement in paragraph (b)(5) of this section may be extended 1 year for each year of full-time laboratory experience obtained before January 1, 1958 required by State law for a laboratory director license. An exception to the July 1, 1971 qualifying date in paragraph (b)(5) of this section was made provided that the individual requested qualification approval by October 21, 1975 and had been employed in a laboratory for at least 3 years of the 5 years preceding the date of submission of his qualifications.
[58 FR 5233, Jan. 19, 1993]
§ 493.1407 Standard; Laboratory director responsibilities.
The laboratory director is responsible for the overall operation and administration of the laboratory, including the employment of personnel who are competent to perform test procedures, and record and report test results promptly, accurate, and proficiently and for assuring compliance with the applicable regulations.
(a) The laboratory director, if qualified, may perform the duties of the technical consultant, clinical consultant, and testing personnel, or delegate these responsibilities to personnel meeting the qualifications of §§ 493.1409, 493.1415, and 493.1421, respectively.
(b) If the laboratory director reapportions performance of his or her responsibilities, he or she remains responsible for ensuring that all duties are properly performed.
(c) The laboratory director must be accessible to the laboratory to provide onsite, telephone or electronic consultation as needed.
(d) Each individual may direct no more than five laboratories.
(e) The laboratory director must—
(1) Ensure that testing systems developed and used for each of the tests performed in the laboratory provide quality laboratory services for all aspects of test performance, which includes the preanalytic, analytic, and postanalytic phases of testing;
(2) Ensure that the physical plant and environmental conditions of the laboratory are appropriate for the testing performed and provide a safe environment in which employees are protected from physical, chemical, and biological hazards;
(3) Ensure that—
(i) The test methodologies selected have the capability of providing the quality of results required for patient care;
(ii) Verification procedures used are adequate to determine the accuracy, precision, and other pertinent performance characteristics of the method; and
(iii) Laboratory personnel are performing the test methods as required for accurate and reliable results;
(4) Ensure that the laboratory is enrolled in an HHS approved proficiency testing program for the testing performed and that—
(i) The proficiency testing samples are tested as required under subpart H of this part;
(ii) The results are returned within the timeframes established by the proficiency testing program;
(iii) All proficiency testing reports received are reviewed by the appropriate staff to evaluate the laboratory's performance and to identify any problems that require corrective action; and
(iv) An approved corrective action plan is followed when any proficiency testing results are found to be unacceptable or unsatisfactory;
(5) Ensure that the quality control and quality assessment programs are established and maintained to assure the quality of laboratory services provided and to identify failures in quality as they occur;
(6) Ensure the establishment and maintenance of acceptable levels of analytical performance for each test system;
(7) Ensure that all necessary remedial actions are taken and documented whenever significant deviations from the laboratory's established performance specifications are identified, and that patient test results are reported only when the system is functioning properly;
(8) Ensure that reports of test results include pertinent information required for interpretation;
(9) Ensure that consultation is available to the laboratory's clients on matters relating to the quality of the test results reported and their interpretation concerning specific patient conditions;
(10) Employ a sufficient number of laboratory personnel with the appropriate education and either experience or training to provide appropriate consultation, properly supervise and accurately perform tests and report test results in accordance with the personnel responsibilities described in this subpart;
(11) Ensure that prior to testing patients' specimens, all personnel have the appropriate education and experience, receive the appropriate training for the type and complexity of the services offered, and have demonstrated that they can perform all testing operations reliably to provide and report accurate results;
(12) Ensure that policies and procedures are established for monitoring individuals who conduct preanalytical, analytical, and postanalytical phases of testing to assure that they are competent and maintain their competency to process specimens, perform test procedures and report test results promptly and proficiently, and whenever necessary, identify needs for remedial training or continuing education to improve skills;
(13) Ensure that an approved procedure manual is available to all personnel responsible for any aspect of the testing process; and
(14) Specify, in writing, the responsibilities and duties of each consultant and each person, engaged in the performance of the preanalytic, analytic, and postanalytic phases of testing, that identifies which examinations and procedures each individual is authorized to perform, whether supervision is required for specimen processing, test performance or results reporting, and whether consultant or director review is required prior to reporting patient test results.
[57 FR 7172, Feb. 28, 1992, as amended at 68 FR 3713, Jan. 24, 2003]
§ 493.1409 Condition: Laboratories performing moderate complexity testing; technical consultant.
The laboratory must have a technical consultant who meets the qualification requirements of § 493.1411 of this subpart and provides technical oversight in accordance with § 493.1413 of this subpart.
§ 493.1411 Standard; Technical consultant qualifications.
The laboratory must employ one or more individuals who are qualified by education and either training or experience to provide technical consultation for each of the specialties and subspecialties of service in which the laboratory performs moderate complexity tests or procedures. The director of a laboratory performing moderate complexity testing may function as the technical consultant provided he or she meets the qualifications specified in this section.
(a) The technical consultant must possess a current license issued by the State in which the laboratory is located, if such licensing is required.
(b) The technical consultant must—
(1)
(i) Be a doctor of medicine or doctor of osteopathy licensed to practice medicine or osteopathy in the State in which the laboratory is located; and
(ii) Be certified in anatomic or clinical pathology, or both, by the American Board of Pathology or the American Osteopathic Board of Pathology or possess qualifications that are equivalent to those required for such certification; or
(2)
(i) Be a doctor of medicine, doctor of osteopathy, or doctor of podiatric medicine licensed to practice medicine, osteopathy, or podiatry in the State in which the laboratory is located; and
(ii) Have at least one year of laboratory training or experience, or both in non-waived testing, in the designated specialty or subspecialty areas of service for which the technical consultant is responsible (for example, physicians certified either in hematology or hematology and medical oncology by the American Board of Internal Medicine are qualified to serve as the technical consultant in hematology); or
(3)
(i) Hold an earned doctoral or master's degree in a chemical, physical, biological or clinical laboratory science or medical technology from an accredited institution; and
(ii) Have at least one year of laboratory training or experience, or both in non-waived testing, in the designated specialty or subspecialty areas of service for which the technical consultant is responsible; or
(4)
(i) Have earned a bachelor's degree in a chemical, physical or biological science or medical technology from an accredited institution; and
(ii) Have at least 2 years of laboratory training or experience, or both in non-waived testing, in the designated specialty or subspecialty areas of service for which the technical consultant is responsible.
The technical consultant requirements for “laboratory training or experience, or both” in each specialty or subspecialty may be acquired concurrently in more than one of the specialties or subspecialties of service, excluding waived tests. For example, an individual who has a bachelor's degree in biology and additionally has documentation of 2 years of work experience performing tests of moderate complexity in all specialties and subspecialties of service, would be qualified as a technical consultant in a laboratory performing moderate complexity testing in all specialties and subspecialties of service.
[57 FR 7172, Feb. 28, 1992, as amended at 58 FR 5234, Jan. 19, 1993]
§ 493.1413 Standard; Technical consultant responsibilities.
The technical consultant is responsible for the technical and scientific oversight of the laboratory. The technical consultant is not required to be onsite at all times testing is performed; however, he or she must be available to the laboratory on an as needed basis to provide consultation, as specified in paragraph (a) of this section.
(a) The technical consultant must be accessible to the laboratory to provide on-site, telephone, or electronic consultation; and
(b) The technical consultant is responsible for—
(1) Selection of test methodology appropriate for the clinical use of the test results;
(2) Verification of the test procedures performed and the establishment of the laboratory's test performance characteristics, including the precision and accuracy of each test and test system;
(3) Enrollment and participation in an HHS approved proficiency testing program commensurate with the services offered;
(4) Establishing a quality control program appropriate for the testing performed and establishing the parameters for acceptable levels of analytic performance and ensuring that these levels are maintained throughout the entire testing process from the initial receipt of the specimen, through sample analysis and reporting of test results;
(5) Resolving technical problems and ensuring that remedial actions are taken whenever test systems deviate from the laboratory's established performance specifications;
(6) Ensuring that patient test results are not reported until all corrective actions have been taken and the test system is functioning properly;
(7) Identifying training needs and assuring that each individual performing tests receives regular in-service training and education appropriate for the type and complexity of the laboratory services performed;
(8) Evaluating the competency of all testing personnel and assuring that the staff maintain their competency to perform test procedures and report test results promptly, accurately and proficiently. The procedures for evaluation of the competency of the staff must include, but are not limited to—
(i) Direct observations of routine patient test performance, including patient preparation, if applicable, specimen handling, processing and testing;
(ii) Monitoring the recording and reporting of test results;
(iii) Review of intermediate test results or worksheets, quality control records, proficiency testing results, and preventive maintenance records;
(iv) Direct observation of performance of instrument maintenance and function checks;
(v) Assessment of test performance through testing previously analyzed specimens, internal blind testing samples or external proficiency testing samples; and
(vi) Assessment of problem solving skills; and
(9) Evaluating and documenting the performance of individuals responsible for moderate complexity testing at least semiannually during the first year the individual tests patient specimens. Thereafter, evaluations must be performed at least annually unless test methodology or instrumentation changes, in which case, prior to reporting patient test results, the individual's performance must be reevaluated to include the use of the new test methodology or instrumentation.
§ 493.1415 Condition: Laboratories performing moderate complexity testing; clinical consultant.
The laboratory must have a clinical consultant who meets the qualification requirements of § 493.1417 of this part and provides clinical consultation in accordance with § 493.1419 of this part.
§ 493.1417 Standard; Clinical consultant qualifications.
The clinical consultant must be qualified to consult with and render opinions to the laboratory's clients concerning the diagnosis, treatment and management of patient care. The clinical consultant must—
(a) Be qualified as a laboratory director under § 493.1405(b) (1), (2), or (3)(i); or
(b) Be a doctor of medicine, doctor of osteopathy or doctor of podiatric medicine and possess a license to practice medicine, osteopathy or podiatry in the State in which the laboratory is located.
[57 FR 7172, Feb. 28, 1992, as amended at 58 FR 5234, Jan. 19, 1993]
§ 493.1419 Standard; Clinical consultant responsibilities.
The clinical consultant provides consultation regarding the appropriateness of the testing ordered and interpretation of test results. The clinical consultant must—
(a) Be available to provide clinical consultation to the laboratory's clients;
(b) Be available to assist the laboratory's clients in ensuring that appropriate tests are ordered to meet the clinical expectations;
(c) Ensure that reports of test results include pertinent information required for specific patient interpretation; and
(d) Ensure that consultation is available and communicated to the laboratory's clients on matters related to the quality of the test results reported and their interpretation concerning specific patient conditions.
§ 493.1421 Condition: Laboratories performing moderate complexity testing; testing personnel.
The laboratory must have a sufficient number of individuals who meet the qualification requirements of § 493.1423, to perform the functions specified in § 493.1425 for the volume and complexity of tests performed.
§ 493.1423 Standard; Testing personnel qualifications.
Each individual performing moderate complexity testing must—
(a) Possess a current license issued by the State in which the laboratory is located, if such licensing is required; and
(b) Meet one of the following requirements:
(1) Be a doctor of medicine or doctor of osteopathy licensed to practice medicine or osteopathy in the State in which the laboratory is located or have earned a doctoral, master's, or bachelor's degree in a chemical, physical, biological or clinical laboratory science, or medical technology from an accredited institution; or
(2) Have earned an associate degree in a chemical, physical or biological science or medical laboratory technology from an accredited institution; or
(3) Be a high school graduate or equivalent and have successfully completed an official military medical laboratory procedures course of at least 50 weeks duration and have held the military enlisted occupational specialty of Medical Laboratory Specialist (Laboratory Technician); or
(4)
(i) Have earned a high school diploma or equivalent; and
(ii) Have documentation of training appropriate for the testing performed prior to analyzing patient specimens. Such training must ensure that the individual has—
(A) The skills required for proper specimen collection, including patient preparation, if applicable, labeling, handling, preservation or fixation, processing or preparation, transportation and storage of specimens;
(B) The skills required for implementing all standard laboratory procedures;
(C) The skills required for performing each test method and for proper instrument use;
(D) The skills required for performing preventive maintenance, troubleshooting and calibration procedures related to each test performed;
(E) A working knowledge of reagent stability and storage;
(F) The skills required to implement the quality control policies and procedures of the laboratory;
(G) An awareness of the factors that influence test results; and
(H) The skills required to assess and verify the validity of patient test results through the evaluation of quality control sample values prior to reporting patient test results.
[57 FR 7172, Feb. 28, 1992, as amended at 58 FR 5234, Jan. 19, 1993]
§ 493.1425 Standard; Testing personnel responsibilities.
The testing personnel are responsible for specimen processing, test performance, and for reporting test results.
(a) Each individual performs only those moderate complexity tests that are authorized by the laboratory director and require a degree of skill commensurate with the individual's education, training or experience, and technical abilities.
(b) Each individual performing moderate complexity testing must—
(1) Follow the laboratory's procedures for specimen handling and processing, test analyses, reporting and maintaining records of patient test results;
(2) Maintain records that demonstrate that proficiency testing samples are tested in the same manner as patient samples;
(3) Adhere to the laboratory's quality control policies, document all quality control activities, instrument and procedural calibrations and maintenance performed;
(4) Follow the laboratory's established corrective action policies and procedures whenever test systems are not within the laboratory's established acceptable levels of performance;
(5) Be capable of identifying problems that may adversely affect test performance or reporting of test results and either must correct the problems or immediately notify the technical consultant, clinical consultant or director; and
(6) Document all corrective actions taken when test systems deviate from the laboratory's established performance specifications.
Laboratories Performing High Complexity Testing
§ 493.1441 Condition: Laboratories performing high complexity testing; laboratory director.
The laboratory must have a director who meets the qualification requirements of § 493.1443 of this subpart and provides overall management and direction in accordance with § 493.1445 of this subpart.
§ 493.1443 Standard; Laboratory director qualifications.
The laboratory director must be qualified to manage and direct the laboratory personnel and performance of high complexity tests and must be eligible to be an operator of a laboratory within the requirements of subpart R.
(a) The laboratory director must possess a current license as a laboratory director issued by the State in which the laboratory is located, if such licensing is required; and
(b) The laboratory director must—
(1)
(i) Be a doctor of medicine or doctor of osteopathy licensed to practice medicine or osteopathy in the State in which the laboratory is located; and
(ii) Be certified in anatomic or clinical pathology, or both, by the American Board of Pathology or the American Osteopathic Board of Pathology or possess qualifications that are equivalent to those required for such certification; or
(2) Be a doctor of medicine, a doctor of osteopathy or doctor of podiatric medicine licensed to practice medicine, osteopathy or podiatry in the State in which the laboratory is located; and
(i) Have at least one year of laboratory training during medical residency (for example, physicians certified either in hematology or hematology and medical oncology by the American Board of Internal Medicine); or
(ii) Have at least 2 years of experience directing or supervising high complexity testing; or
(3) Hold an earned doctoral degree in a chemical, physical, biological, or clinical laboratory science from an accredited institution and—
(i) Be certified and continue to be certified by a board approved by HHS; or
(ii) Before February 24, 2003, must have served or be serving as a director of a laboratory performing high complexity testing and must have at least—
(A) Two years of laboratory training or experience, or both; and
(B) Two years of laboratory experience directing or supervising high complexity testing.
(4) Be serving as a laboratory director and must have previously qualified or could have qualified as a laboratory director under regulations at 42 CFR 493.1415, published March 14, 1990 at 55 FR 9538, on or before February 28, 1992; or
(5) On or before February 28, 1992, be qualified under State law to direct a laboratory in the State in which the laboratory is located; or
(6) For the subspecialty of oral pathology, be certified by the American Board of Oral Pathology, American Board of Pathology, the American Osteopathic Board of Pathology, or possess qualifications that are equivalent to those required for certification.
[57 FR 7172, Feb. 28, 1992, as amended at 58 FR 5234, Jan. 19, 1993; 59 FR 62609, Dec. 6, 1994; 62 FR 25858, May 12, 1997; 63 FR 55034, Oct. 14, 1998; 65 FR 82944, Dec. 29, 2000; 68 FR 3713, Jan. 24, 2003]
§ 493.1445 Standard; Laboratory director responsibilities.
The laboratory director is responsible for the overall operation and administration of the laboratory, including the employment of personnel who are competent to perform test procedures, record and report test results promptly, accurately and proficiently, and for assuring compliance with the applicable regulations.
(a) The laboratory director, if qualified, may perform the duties of the technical supervisor, clinical consultant, general supervisor, and testing personnel, or delegate these responsibilities to personnel meeting the qualifications under §§ 493.1447, 493.1453, 493.1459, and 493.1487, respectively.
(b) If the laboratory director reapportions performance of his or her responsibilities, he or she remains responsible for ensuring that all duties are properly performed.
(c) The laboratory director must be accessible to the laboratory to provide onsite, telephone or electronic consultation as needed.
(d) Each individual may direct no more than five laboratories.
(e) The laboratory director must—
(1) Ensure that testing systems developed and used for each of the tests performed in the laboratory provide quality laboratory services for all aspects of test performance, which includes the preanalytic, analytic, and postanalytic phases of testing;
(2) Ensure that the physical plant and environmental conditions of the laboratory are appropriate for the testing performed and provide a safe environment in which employees are protected from physical, chemical, and biological hazards;
(3) Ensure that—
(i) The test methodologies selected have the capability of providing the quality of results required for patient care;
(ii) Verification procedures used are adequate to determine the accuracy, precision, and other pertinent performance characteristics of the method; and
(iii) Laboratory personnel are performing the test methods as required for accurate and reliable results;
(4) Ensure that the laboratory is enrolled in an HHS-approved proficiency testing program for the testing performed and that—
(i) The proficiency testing samples are tested as required under subpart H of this part;
(ii) The results are returned within the timeframes established by the proficiency testing program;
(iii) All proficiency testing reports received are reviewed by the appropriate staff to evaluate the laboratory's performance and to identify any problems that require corrective action; and
(iv) An approved corrective action plan is followed when any proficiency testing result is found to be unacceptable or unsatisfactory;
(5) Ensure that the quality control and quality assessment programs are established and maintained to assure the quality of laboratory services provided and to identify failures in quality as they occur;
(6) Ensure the establishment and maintenance of acceptable levels of analytical performance for each test system;
(7) Ensure that all necessary remedial actions are taken and documented whenever significant deviations from the laboratory's established performance characteristics are identified, and that patient test results are reported only when the system is functioning properly;
(8) Ensure that reports of test results include pertinent information required for interpretation;
(9) Ensure that consultation is available to the laboratory's clients on matters relating to the quality of the test results reported and their interpretation concerning specific patient conditions;
(10) Ensure that a general supervisor provides on-site supervision of high complexity test performance by testing personnel qualified under § 493.1489(b)(4);
(11) Employ a sufficient number of laboratory personnel with the appropriate education and either experience or training to provide appropriate consultation, properly supervise and accurately perform tests and report test results in accordance with the personnel responsibilities described in this subpart;
(12) Ensure that prior to testing patients' specimens, all personnel have the appropriate education and experience, receive the appropriate training for the type and complexity of the services offered, and have demonstrated that they can perform all testing operations reliably to provide and report accurate results;
(13) Ensure that policies and procedures are established for monitoring individuals who conduct preanalytical, analytical, and postanalytical phases of testing to assure that they are competent and maintain their competency to process specimens, perform test procedures and report test results promptly and proficiently, and whenever necessary, identify needs for remedial training or continuing education to improve skills;
(14) Ensure that an approved procedure manual is available to all personnel responsible for any aspect of the testing process; and
(15) Specify, in writing, the responsibilities and duties of each consultant and each supervisor, as well as each person engaged in the performance of the preanalytic, analytic, and postanalytic phases of testing, that identifies which examinations and procedures each individual is authorized to perform, whether supervision is required for specimen processing, test performance or result reporting and whether supervisory or director review is required prior to reporting patient test results.
[57 FR 7172, Feb. 28, 1992, as amended at 68 FR 3714, Jan. 24, 2003]
§ 493.1447 Condition: Laboratories performing high complexity testing; technical supervisor.
The laboratory must have a technical supervisor who meets the qualification requirements of § 493.1449 of this subpart and provides technical supervision in accordance with § 493.1451 of this subpart.
§ 493.1449 Standard; Technical supervisor qualifications.
The laboratory must employ one or more individuals who are qualified by education and either training or experience to provide technical supervision for each of the specialties and subspecialties of service in which the laboratory performs high complexity tests or procedures. The director of a laboratory performing high complexity testing may function as the technical supervisor provided he or she meets the qualifications specified in this section.
(a) The technical supervisor must possess a current license issued by the State in which the laboratory is located, if such licensing is required; and
(b) The laboratory may perform anatomic and clinical laboratory procedures and tests in all specialties and subspecialties of services except histocompatibility and clinical cytogenetics services provided the individual functioning as the technical supervisor—
(1) Is a doctor of medicine or doctor of osteopathy licensed to practice medicine or osteopathy in the State in which the laboratory is located; and
(2) Is certified in both anatomic and clinical pathology by the American Board of Pathology or the American Osteopathic Board of Pathology or Possesses qualifications that are equivalent to those required for such certification.
(c) If the requirements of paragraph (b) of this section are not met and the laboratory performs tests in the subspecialty of bacteriology, the individual functioning as the technical supervisor must—
(1)
(i) Be a doctor of medicine or doctor of osteopathy licensed to practice medicine or osteopathy in the State in which the laboratory is located; and
(ii) Be certified in clinical pathology by the American Board of Pathology or the American Osteopathic Board of Pathology or possess qualifications that are equivalent to those required for such certification; or
(2)
(i) Be a doctor of medicine, doctor of osteopathy, or doctor of podiatric medicine licensed to practice medicine, osteopathy, or podiatry in the State in which the laboratory is located; and
(ii) Have at least one year of laboratory training or experience, or both, in high complexity testing within the specialty of microbiology with a minimum of 6 months experience in high complexity testing within the subspecialty of bacteriology; or
(3)
(i) Have an earned doctoral degree in a chemical, physical, biological or clinical laboratory science from an accredited institution; and
(ii) Have at least 1 year of laboratory training or experience, or both, in high complexity testing within the specialty of microbiology with a minimum of 6 months experience in high complexity testing within the subspecialty of bacteriology; or
(4)
(i) Have earned a master's degree in a chemical, physical, biological or clinical laboratory science or medical technology from an accredited institution; and
(ii) Have at least 2 years of laboratory training or experience, or both, in high complexity testing within the specialty of microbiology with a minimum of 6 months experience in high complexity testing within the subspecialty of bacteriology; or
(5)
(i) Have earned a bachelor's degree in a chemical, physical, or biological science or medical technology from an accredited institution; and
(ii) Have at least 4 years of laboratory training or experience, or both, in high complexity testing within the specialty of microbiology with a minimum of 6 months experience in high complexity testing within the subspecialty of bacteriology.
(d) If the requirements of paragraph (b) of this section are not met and the laboratory performs tests in the subspecialty of mycobacteriology, the individual functioning as the technical supervisor must—
(1)
(i) Be a doctor of medicine or doctor of osteopathy licensed to practice medicine or osteopathy in the State in which the laboratory is located; and
(ii) Be certified in clinical pathology by the American Board of Pathology or the American Osteopathic Board of Pathology or possess qualifications that are equivalent to those required for such certification; or
(2)
(i) Be a doctor of medicine, doctor of osteopathy, or doctor or podiatric medicine licensed to practice medicine, osteopathy, or podiatry in the State in which the laboratory is located; and
(ii) Have at least 1 year of laboratory training or experience, or both, in high complexity testing within the specialty of microbiology with a minimum of 6 months experience in high complexity testing within the subspecialty of mycobacteriology; or
(3)
(i) Have an earned doctoral degree in a chemical, physical, biological or clinical laboratory science from an accredited institution; and
(ii) Have at least 1 year of laboratory training or experience, or both, in high complexity testing within the specialty of microbiology with a minimum of 6 months experience in high complexity testing within the subspecialty of mycobacteriology; or
(4)
(i) Have earned a master's degree in a chemical, physical, biological or clinical laboratory science or medical technology from an accredited institution; and
(ii) Have at least 2 years of laboratory training or experience, or both, in high complexity testing within the specialty of microbiology with a minimum of 6 months experience in high complexity testing within the subspecialty of mycobacteriology; or
(5)
(i) Have earned a bachelor's degree in a chemical, physical or biological science or medical technology from an accredited institution; and
(ii) Have at least 4 years of laboratory training or experience, or both, in high complexity testing within the specialty of microbiology with a minimum of 6 months experience in high complexity testing within the subspecialty of mycobacteriology.
(e) If the requirements of paragraph (b) of this section are not met and the laboratory performs tests in the subspecialty of mycology, the individual functioning as the technical supervisor must—
(1)
(i) Be a doctor of medicine or doctor of osteopathy licensed to practice medicine or osteopathy in the State in which the laboratory is located; and
(ii) Be certified in clinical pathology by the American Board of Pathology or the American osteopathic Board of Pathology or possess qualifications that are equivalent to those required for such certification; or
(2)
(i) Be a doctor of medicine, doctor of osteopathy, or doctor of podiatric medicine licensed to practice medicine, osteopathy, or podiatry in the State in which the laboratory is located; and
(ii) Have at least 1 year of laboratory training or experience, or both, in high complexity testing within the specialty of microbiology with a minimum of 6 months experience in high complexity testing within the subspecialty of mycology; or
(3)
(i) Have an earned doctoral degree in a chemical, physical, biological or clinical laboratory science from an accredited institution; and
(ii) Have at least 1 year of laboratory training or experience, or both in high complexity testing within the speciality of microbiology with a minimum of 6 months experience in high complexity testing within the subspecialty of mycology; or
(4)
(i) Have earned a master's degree in a chemical, physical, biological or clinical laboratory science or medical technology from an accredited institution; and
(ii) Have at least 2 years of laboratory training or experience, or both, in high complexity testing within the specialty of microbiology with a minimum of 6 months experience in high complexity testing within the subspecialty of mycology; or
(5)
(i) Have earned a bachelor's degree in a chemical, physical or biological science or medical technology from an accredited institution; and
(ii) Have at least 4 years of laboratory training or experience, or both, in high complexity testing within the specialty of microbiology with a minimum of 6 months experience in high complexity testing within the subspecialty of mycology.
(f) If the requirements of paragraph (b) of this section are not met and the laboratory performs tests in the subspecialty of parasitology, the individual functioning as the technical supervisor must—
(1)
(i) Be a doctor of medicine or a doctor of osteopathy licensed to practice medicine or osteopathy in the State in which the laboratory is located; and
(ii) Be certified in clinical pathology by the American Board of Pathology or the American Osteopathic Board of Pathology or possess qualifications that are equivalent to those required for such certification; or
(2)
(i) Be a doctor of medicine, doctor of osteopathy, or doctor of podiatric medicine licensed to practice medicine, osteopathy, or podiatry in the State in which the laboratory is located; and
(ii) Have at least one year of laboratory training or experience, or both, in high complexity testing within the specialty of microbiology with a minimum of 6 months experience in high complexity testing within the subspecialty of parasitology;
(3)
(i) Have an earned doctoral degree in a chemical, physical, biological or clinical laboratory science from an accredited institution; and
(ii) Have at least 1 year of laboratory training or experience, or both, in high complexity testing within the specialty of microbiology with a minimum of 6 months experience in high complexity testing within the subspecialty of parasitology; or
(4)
(i) Have earned a master's degree in a chemical, physical, biological or clinical laboratory science or medical technology from an accredited institution; and
(ii) Have at least 2 years of laboratory training or experience, or both, in high complexity testing within the specialty of microbiology with a minimum of 6 months experience in high complexity testing within the subspecialty of parasitology; or
(5)
(i) Have earned a bachelor's degree in a chemical, physical or biological science or medical technology from an accredited institution; and
(ii) Have at least 4 years of laboratory training or experience, or both, in high complexity testing within the specialty of microbiology with a minimum of 6 months experience in high complexity testing within the subspecialty of parasitology.
(g) If the requirements of paragraph (b) of this section are not met and the laboratory performs tests in the subspecialty of virology, the individual functioning as the technical supervisor must—
(1)
(i) Be a doctor of medicine or doctor of osteopathy licensed to practice medicine or osteopathy in the State in which the laboratory is located; and
(ii) Be certified in clinical pathology by the American Board of Pathology or the American Osteopathic Board of Pathology or possess qualifications that are equivalent to those required for such certification; or
(2)
(i) Be a doctor of medicine, doctor of osteopathy, or doctor of podiatric medicine licensed to practice medicine, osteopathy, or podiatry in the State in which the laboratory is located; and
(ii) Have at least 1 year of laboratory training or experience, or both, in high complexity testing within the specialty of microbiology with a minimum of 6 months experience in high complexity testing within the subspecialty of virology; or
(3)
(i) Have an earned doctoral degree in a chemical, physical, biological or clinical laboratory science from an accredited institution; and
(ii) Have at least 1 year of laboratory training or experience, or both, in high complexity testing within the specialty of microbiology with a minimum of 6 months experience in high complexity testing within the subspecialty of virology; or
(4)
(i) Have earned a master's degree in a chemical, physical, biological or clinical laboratory science or medical technology from an accredited institution; and
(ii) Have at least 2 years of laboratory training or experience, or both, in high complexity testing within the specialty of microbiology with a minimum of 6 months experience in high complexity testing within the subspecialty of virology; or
(5)
(i) Have earned a bachelor's degree in a chemical, physical or biological science or medical technology from an accredited institution; and
(ii) Have at least 4 years of laboratory training or experience, or both, in high complexity testing within the specialty of microbiology with a minimum of 6 months experience in high complexity testing within the subspecialty of virology.
(h) If the requirements of paragraph (b) of this section are not met and the laboratory performs tests in the specialty of diagnostic immunology, the individual functioning as the technical supervisor must—
(1)
(i) Be a doctor of medicine or a doctor of osteopathy licensed to practice medicine or osteopathy in the State in which the laboratory is located; and
(ii) Be certified in clinical pathology by the American Board of Pathology or the American Osteopathic Board of Pathology or possess qualifications that are equivalent to those required for such certification; or
(2)
(i) Be a doctor of medicine, doctor of osteopathy, or doctor of podiatric medicine licensed to practice medicine, osteopathy, or podiatry in the State in which the laboratory is located; and
(ii) Have at least 1 year of laboratory training or experience, or both, in high complexity testing for the specialty of diagnostic immunology; or
(3)
(i) Have an earned doctoral degree in a chemical, physical, biological or clinical laboratory science from an accredited institution; and
(ii) Have at least 1 year of laboratory training or experience, or both, in high complexity testing within the specialty of diagnostic immunology; or
(4)
(i) Have earned a master's degree in a chemical, physical, biological or clinical laboratory science or medical technology from an accredited institution; and
(ii) Have at least 2 years of laboratory training or experience, or both, in high complexity testing for the specialty of diagnostic immunology; or
(5)
(i) Have earned a bachelor's degree in a chemical, physical or biological science or medical technology from an accredited institution; and
(ii) Have at least 4 years of laboratory training or experience, or both, in high complexity testing for the specialty of diagnostic immunology.
(i) If the requirements of paragraph (b) of this section are not met and the laboratory performs tests in the specialty of chemistry, the individual functioning as the technical supervisor must—
(1)
(i) Be a doctor of medicine or doctor of osteopathy licensed to practice medicine or osteopathy in the State in which the laboratory is located; and
(ii) Be certified in clinical pathology by the American Board of Pathology or the American Osteopathic Board of Pathology or possess qualifications that are equivalent to those required for such certification; or
(2)
(i) Be a doctor of medicine, doctor of osteopathy, or doctor of podiatric medicine licensed to practice medicine, osteopathy, or podiatry in the State in which the laboratory is located; and
(ii) Have at least 1 year of laboratory training or experience, or both, in high complexity testing for the specialty of chemistry; or
(3)
(i) Have an earned doctoral degree in a chemical, physical, biological or clinical laboratory science from an accredited institution; and
(ii) Have at least 1 year of laboratory training or experience, or both, in high complexity testing within the specialty of chemistry; or
(4)
(i) Have earned a master's degree in a chemical, physical, biological or clinical laboratory science or medical technology from an accredited institution; and
(ii) Have at least 2 years of laboratory training or experience, or both, in high complexity testing for the specialty of chemistry; or
(5)
(i) Have earned a bachelor's degree in a chemical, physical or biological science or medical technology from an accredited institution; and
(ii) Have at least 4 years of laboratory training or experience, or both, in high complexity testing for the specialty of chemistry.
(j) If the requirements of paragraph (b) of this section are not met and the laboratory performs tests in the specialty of hematology, the individual functioning as the technical supervisor must—
(1)
(i) Be a doctor of medicine or a doctor of osteopathy licensed to practice medicine or osteopathy in the State in which the laboratory is located; and
(ii) Be certified in clinical pathology by the American Board of Pathology or the American Osteopathic Board of Pathology or possess qualifications that are equivalent to those required for such certification; or
(2)
(i) Be a doctor of medicine, doctor of osteopathy, or doctor of podiatric medicine licensed to practice medicine, osteopathy, or podiatry in the State in which the laboratory is located; and
(ii) Have at least one year of laboratory training or experience, or both, in high complexity testing for the specialty of hematology (for example, physicians certified either in hematology or hematology and medical oncology by the American Board of Internal Medicine); or
(3)
(i) Have an earned doctoral degree in a chemical, physical, biological or clinical laboratory science from an accredited institution; and
(ii) Have at least 1 year of laboratory training or experience, or both, in high complexity testing within the specialty of hematology; or
(4)
(i) Have earned a master's degree in a chemical, physical, biological or clinical laboratory science or medical technology from an accredited institution; and
(ii) Have at least 2 years of laboratory training or experience, or both, in high complexity testing for the specialty of hematology; or
(5)
(i) Have earned a bachelor's degree in a chemical, physical or biological science or medical technology from an accredited institution; and
(ii) Have at least 4 years of laboratory training or experience, or both, in high complexity testing for the specialty of hematology.
(k)
(1) If the requirements of paragraph (b) of this section are not met and the laboratory performs tests in the subspecialty of cytology, the individual functioning as the technical supervisor must—
(i) Be a doctor of medicine or a doctor of osteopathy licensed to practice medicine or osteopathy in the State in which the laboratory is located; and
(ii) Meet one of the following requirements—
(A) Be certified in anatomic pathology by the American Board of Pathology or the American Osteopathic Board of Pathology or possess qualifications that are equivalent to those required for such certification; or
(B) Be certified by the American Society of Cytology to practice cytopathology or possess qualifications that are equivalent to those required for such certification;
(2) An individual qualified under § 493.1449(b) or paragraph (k)(1) of this section may delegate some of the cytology technical supervisor responsibilities to an individual who is in the final year of full-time training leading to certification specified in paragraphs (b) or (k)(1)(ii)(A) of this section provided the technical supervisor qualified under § 493.1449(b) or paragraph (k)(1) of this section remains ultimately responsible for ensuring that all of the responsibilities of the cytology technical supervisor are met.
(l) If the requirements of paragraph (b) of this section are not met and the laboratory performs tests in the subspecialty of histopathology, the individual functioning as the technical supervisor must—
(1) Meet one of the following requirements:
(i)
(A) Be a doctor of medicine or a doctor of osteopathy licensed to practice medicine or osteopathy in the State in which the laboratory is located; and
(B) Be certified in anatomic pathology by the American Board of Pathology or the American Osteopathic Board of Pathology or possess qualifications that are equivalent to those required for such certification;
(ii) An individual qualified under § 493.1449(b) or paragraph (l)(1) of this section may delegate to an individual who is a resident in a training program leading to certification specified in paragraph (b) or (l)(1)(i)(B) of this section, the responsibility for examination and interpretation of histopathology specimens.
(2) For tests in dermatopathology, meet one of the following requirements:
(i)
(A) Be a doctor of medicine or doctor of osteopathy licensed to practice medicine or osteopathy in the State in which the laboratory is located and—
(B) Meet one of the following requirements:
(1) Be certified in anatomic pathology by the American Board of Pathology or the American Osteopathic Board of Pathology or possess qualifications that are equivalent to those required for such certification; or
(2) Be certified in dermatopathology by the American Board of Dermatology and the American Board of Pathology or possess qualifications that are equivalent to those required for such certification; or
(3) Be certified in dermatology by the American Board of Dermatology or possess qualifications that are equivalent to those required for such certification; or
(ii) An individual qualified under § 493.1449(b) or paragraph (l)(2)(i) of this section may delegate to an individual who is a resident in a training program leading to certification specified in paragraphs (b) or (l)(2)(i)(B) of this section, the responsibility for examination and interpretation of dermatopathology specimens.
(3) For tests in ophthalmic pathology, meet one of the following requirements:
(i)
(A) Be a doctor of medicine or doctor of osteopathy licensed to practice medicine or osteopathy in the State in which the laboratory is located and—
(B) Must meet one of the following requirements:
(1) Be certified in anatomic pathology by the American Board of Pathology or the American Osteopathic Board of Pathology or possess qualifications that are equivalent to those required for such certification; or
(2) Be certified by the American Board of Ophthalmology or possess qualifications that are equivalent to those required for such certitication and have successfully completed at least 1 year of formal post-residency fellowship training in ophthalmic pathology; or
(ii) An individual qualified under § 493.1449(b) or paragraph (1)(3)(i) of this section may delegate to an individual who is a resident in a training program leading to certification specified in paragraphs (b) or (1)(3)(i)(B) of this section, the responsibility for examination and interpretation of ophthalmic specimens; or
(m) If the requirements of paragraph (b) of this section are not met and the laboratory performs tests in the subspecialty of oral pathology, the individual functioning as the technical supervisor must meet one of the following requirements:
(1)
(i) Be a doctor of medicine or a doctor of osteopathy licensed to practice medicine or osteopathy in the State in which the laboratory is located and—
(ii) Be certified in anatomic pathology by the American Board of Pathology or the American Osteopathic Board of Pathology or possess qualifications that are equivalent to those required for such certification; or
(2) Be certified in oral pathology by the American Board of Oral Pathology or possess qualifications for such certification; or
(3) An individual qualified under § 493.1449(b) or paragraph (m) (1) or (2) of this section may delegate to an individual who is a resident in a training program leading to certification specified in paragraphs (b) or (m) (1) or (2) of this section, the responsibility for examination and interpretation of oral pathology specimens.
(n) If the requirements of paragraph (b) of this section are not met and the laboratory performs tests in the specialty of radiobioassay, the individual functioning as the technical supervisor must—
(1)
(i) Be a doctor of medicine or a doctor of osteopathy licensed to practice medicine or osteopathy in the State in which the laboratory is located; and
(ii) Be certified in clinical pathology by the American Board of Pathology or the American Osteopathic Board of Pathology or possess qualifications that are equivalent to those required for such certification; or
(2)
(i) Be a doctor of medicine, doctor of osteopathy, or doctor of podiatric medicine licensed to practice medicine, osteopathy, or podiatry in the State in which the laboratory is located; and
(ii) Have at least 1 year of laboratory training or experience, or both, in high complexity testing for the specialty of radiobioassay; or
(3)
(i) Have an earned doctoral degree in a chemical, physical, biological or clinical laboratory science from an accredited institution; and
(ii) Have at least 1 year of laboratory training or experience, or both, in high complexity testing within the specialty of radiobioassay; or
(4)
(i) Have earned a master's degree in a chemical, physical, biological or clinical laboratory science or medical technology from an accredited institution; and
(ii) Have at least 2 years of laboratory training or experience, or both, in high complexity testing for the specialty of radiobioassay; or
(5)
(i) Have earned a bachelor's degree in a chemical, physical or biological science or medical technology from an accredited institution; and
(ii) Have at least 4 years of laboratory training or experience, or both, in high complexity testing for the specialty of radiobioassay.
(o) If the laboratory performs tests in the specialty of histocompatibility, the individual functioning as the technical supervisor must either—
(1)
(i) Be a doctor of medicine, doctor of osteopathy, or doctor of podiatric medicine licensed to practice medicine, osteopathy, or podiatry in the State in which the laboratory is located; and
(ii) Have training or experience that meets one of the following requirements:
(A) Have 4 years of laboratory training or experience, or both, within the specialty of histocompatibility; or
(B)
(1) Have 2 years of laboratory training or experience, or both, in the specialty of general immunology; and
(2) Have 2 years of laboratory training or experience, or both, in the specialty of histocompatibility; or
(2)
(i) Have an earned doctoral degree in a biological or clinical laboratory science from an accredited institution; and
(ii) Have training or experience that meets one of the following requirements:
(A) Have 4 years of laboratory training or experience, or both, within the specialty of histocompatibility; or
(B)
(1) Have 2 years of laboratory training or experience, or both, in the specialty of general immunology; and
(2) Have 2 years of laboratory training or experience, or both, in the specialty of histocompatibility.
(p) If the laboratory performs tests in the specialty of clinical cytogenetics, the individual functioning as the technical supervisor must—
(1)
(i) Be a doctor of medicine, doctor of osteopathy, or doctor of podiatric medicine licensed to practice medicine, osteopathy, or podiatry in the State in which the laboratory is located; and
(ii) Have 4 years of training or experience, or both, in genetics, 2 of which have been in clinical cytogenetics; or
(2)
(i) Hold an earned doctoral degree in a biological science, including biochemistry, or clinical laboratory science from an accredited institution; and
(ii) Have 4 years of training or experience, or both, in genetics, 2 of which have been in clinical cytogenetics.
(q) If the requirements of paragraph (b) of this section are not met and the laboratory performs tests in the specialty of immunohematology, the individual functioning as the technical supervisor must—
(1)
(i) Be a doctor of medicine or a doctor of osteopathy licensed to practice medicine or osteopathy in the State in which the laboratory is located; and
(ii) Be certified in clinical pathology by the American Board of Pathology or the American Osteopathic Board of Pathology or possess qualifications that are equivalent to those required for such certification; or
(2)
(i) Be a doctor of medicine, doctor of osteopathy, or doctor of podiatric medicine licensed to practice medicine, osteopathy, or podiatry in the State in which the laboratory is located; and
(ii) Have at least one year of laboratory training or experience, or both, in high complexity testing for the specialty of immunohematology.
The technical supervisor requirements for “laboratory training or experience, or both” in each specialty or subspecialty may be acquired concurrently in more than one of the specialties or subspecialties of service. For example, an individual, who has a doctoral degree in chemistry and additionally has documentation of 1 year of laboratory experience working concurrently in high complexity testing in the specialties of microbiology and chemistry and 6 months of that work experience included high complexity testing in bacteriology, mycology, and mycobacteriology, would qualify as the technical supervisor for the specialty of chemistry and the subspecialties of bacteriology, mycology, and mycobacteriology.
[57 FR 7172, Feb. 28, 1992, as amended at 58 FR 5234, Jan. 19, 1993]
§ 493.1451 Standard: Technical supervisor responsibilities.
The technical supervisor is responsible for the technical and scientific oversight of the laboratory. The technical supervisor is not required to be on site at all times testing is performed; however, he or she must be available to the laboratory on an as needed basis to provide supervision as specified in (a) of this section.
(a) The technical supervisor must be accessible to the laboratory to provide on-site, telephone, or electronic consultation; and
(b) The technical supervisor is responsible for—
(1) Selection of the test methodology that is appropriate for the clinical use of the test results;
(2) Verification of the test procedures performed and establishment of the laboratory's test performance characteristics, including the precision and accuracy of each test and test system;
(3) Enrollment and participation in an HHS approved proficiency testing program commensurate with the services offered;
(4) Establishing a quality control program appropriate for the testing performed and establishing the parameters for acceptable levels of analytic performance and ensuring that these levels are maintained throughout the entire testing process from the initial receipt of the specimen, through sample analysis and reporting of test results;
(5) Resolving technical problems and ensuring that remedial actions are taken whenever test systems deviate from the laboratory's established performance specifications;
(6) Ensuring that patient test results are not reported until all corrective actions have been taken and the test system is functioning properly;
(7) Identifying training needs and assuring that each individual performing tests receives regular in-service training and education appropriate for the type and complexity of the laboratory services performed;
(8) Evaluating the competency of all testing personnel and assuring that the staff maintain their competency to perform test procedures and report test results promptly, accurately and proficiently. The procedures for evaluation of the competency of the staff must include, but are not limited to—
(i) Direct observations of routine patient test performance, including patient preparation, if applicable, specimen handling, processing and testing;
(ii) Monitoring the recording and reporting of test results;
(iii) Review of intermediate test results or worksheets, quality control records, proficiency testing results, and preventive maintenance records;
(iv) Direct observation of performance of instrument maintenance and function checks;
(v) Assessment of test performance through testing previously analyzed specimens, internal blind testing samples or external proficiency testing samples; and
(vi) Assessment of problem solving skills; and
(9) Evaluating and documenting the performance of individuals responsible for high complexity testing at least semiannually during the first year the individual tests patient specimens. Thereafter, evaluations must be performed at least annually unless test methodology or instrumentation changes, in which case, prior to reporting patient test results, the individual's performance must be reevaluated to include the use of the new test methodology or instrumentation.
(c) In cytology, the technical supervisor or the individual qualified under § 493.1449(k)(2)—
(1) May perform the duties of the cytology general supervisor and the cytotechnologist, as specified in §§ 493.1471 and 493.1485, respectively;
(2) Must establish the workload limit for each individual examining slides;
(3) Must reassess the workload limit for each individual examining slides at least every 6 months and adjust as necessary;
(4) Must perform the functions specified in § 493.1274(d) and (e);
(5) Must ensure that each individual examining gynecologic preparations participates in an HHS approved cytology proficiency testing program, as specified in § 493.945 and achieves a passing score, as specified in § 493.855; and
(6) If responsible for screening cytology slide preparations, must document the number of cytology slides screened in 24 hours and the number of hours devoted during each 24-hour period to screening cytology slides.
[57 FR 7172, Feb. 28, 1992, as amended at 58 FR 5235, Jan. 19, 1993; 68 FR 3714, Jan. 24, 2003]
§ 493.1453 Condition: Laboratories performing high complexity testing; clinical consultant.
The laboratory must have a clinical consultant who meets the requirements of § 493.1455 of this subpart and provides clinical consultation in accordance with § 493.1457 of this subpart.
§ 493.1455 Standard; Clinical consultant qualifications.
The clinical consultant must be qualified to consult with and render opinions to the laboratory's clients concerning the diagnosis, treatment and management of patient care. The clinical consultant must—
(a) Be qualified as a laboratory director under § 493.1443(b)(1), (2), or (3)(i) or, for the subspecialty of oral pathology, § 493.1443(b)(6); or
(b) Be a doctor of medicine, doctor of osteopathy, doctor of podiatric medicine licensed to practice medicine, osteopathy, or podiatry in the State in which the laboratory is located.
[57 FR 7172, Feb. 28, 1992, as amended at 58 FR 5235, Jan. 19, 1993]
§ 493.1457 Standard; Clinical consultant responsibilities.
The clinical consultant provides consultation regarding the appropriateness of the testing ordered and interpretation of test results. The clinical consultant must—
(a) Be available to provide consultation to the laboratory's clients;
(b) Be available to assist the laboratory's clients in ensuring that appropriate tests are ordered to meet the clinical expectations;
(c) Ensure that reports of test results include pertinent information required for specific patient interpretation; and
(d) Ensure that consultation is available and communicated to the laboratory's clients on matters related to the quality of the test results reported and their interpretation concerning specific patient conditions.
§ 493.1459 Condition: Laboratories performing high complexity testing; general supervisor.
The laboratory must have one or more general supervisors who are qualified under § 493.1461 of this subpart to provide general supervision in accordance with § 493.1463 of this subpart.
§ 493.1461 Standard: General supervisor qualifications.
The laboratory must have one or more general supervisors who, under the direction of the laboratory director and supervision of the technical supervisor, provides day-to-day supervision of testing personnel and reporting of test results. In the absence of the director and technical supervisor, the general supervisor must be responsible for the proper performance of all laboratory procedures and reporting of test results.
(a) The general supervisor must possess a current license issued by the State in which the laboratory is located, if such licensing is required; and
(b) The general supervisor must be qualified as a—
(1) Laboratory director under § 493.1443; or
(2) Technical supervisor under § 493.1449.
(c) If the requirements of paragraph (b)(1) or paragraph (b)(2) of this section are not met, the individual functioning as the general supervisor must—
(1)
(i) Be a doctor of medicine, doctor of osteopathy, or doctor of podiatric medicine licensed to practice medicine, osteopathy, or podiatry in the State in which the laboratory is located or have earned a doctoral, master's, or bachelor's degree in a chemical, physical, biological or clinical laboratory science, or medical technology from an accredited institution; and
(ii) Have at least 1 year of laboratory training or experience, or both, in high complexity testing; or
(2)
(i) Qualify as testing personnel under § 493.1489(b)(2); and
(ii) Have at least 2 years of laboratory training or experience, or both, in high complexity testing; or
(3)
(i) Except as specified in paragraph (3)(ii) of this section, have previously qualified as a general supervisor under § 493.1462 on or before February 28, 1992.
(ii) Exception. An individual who achieved a satisfactory grade in a proficiency examination for technologist given by HHS between March 1, 1986 and December 31, 1987, qualifies as a general supervisor if he or she meets the requirements of § 493.1462 on or before January 1, 1994.”
(4) On or before September 1, 1992, have served as a general supervisor of high complexity testing and as of April 24, 1995—
(i) Meet one of the following requirements:
(A) Have graduated from a medical laboratory or clinical laboratory training program approved or accredited by the Accrediting Bureau of Health Education Schools (ABHES), the Commission on Allied Health Education Accreditation (CAHEA), or other organization approved by HHS.
(B) Be a high school graduate or equivalent and have successfully completed an official U.S. military medical laboratory procedures course of at least 50 weeks duration and have held the military enlisted occupational specialty of Medical Laboratory Specialist (Laboratory Technician).
(ii) Have at least 2 years of clinical laboratory training, or experience, or both, in high complexity testing; or
(5) On or before September 1, 1992, have served as a general supervisor of high complexity testing and—
(i) Be a high school graduate or equivalent; and
(ii) Have had at least 10 years of laboratory training or experience, or both, in high complexity testing, including at least 6 years of supervisory experience between September 1, 1982 and September 1, 1992.
(d) For blood gas analysis, the individual providing general supervision must—
(1) Be qualified under § 493.1461(b) (1) or (2), or § 493.1461(c); or
(2)
(i) Have earned a bachelor's degree in respiratory therapy or cardiovascular technology from an accredited institution; and
(ii) Have at least one year of laboratory training or experience, or both, in blood gas analysis; or
(3)
(i) Have earned an associate degree related to pulmonary function from an accredited institution; and
(ii) Have at least two years of training or experience, or both in blood gas analysis.
(e) The general supervisor requirement is met in histopathology, oral pathology, dermatopathology, and ophthalmic pathology because all tests and examinations, must be performed:
(1) In histopathology, by an individual who is qualified as a technical supervisor under § 493.1449(b) or § 493.1449(l)(1);
(2) In dermatopathology, by an individual who is qualified as a technical supervisor under § 493.1449(b) or § 493.1449(l) or (2);
(3) In ophthalmic pathology, by an individual who is qualified as a technical supervisor under § 493.1449(b) or § 493.1449(1)(3); and
(4) In oral pathology, by an individual who is qualified as a technical supervisor under § 493.1449(b) or § 493.1449(m).
[57 FR 7172, Feb. 28, 1992, as amended at 58 FR 5235, Jan. 19, 1993; 58 FR 39155, July 22, 1993; 60 FR 20049, Apr. 24, 1995]
§ 493.1462 General supervisor qualifications on or before February 28, 1992.
To qualify as a general supervisor under § 493.1461(c)(3), an individual must have met or could have met the following qualifications as they were in effect on or before February 28, 1992.
(a) Each supervisor possesses a current license as a laboratory supervisor issued by the State, if such licensing exists; and
(b) The laboratory supervisor—
(1) Who qualifies as a laboratory director under § 493.1406(b)(1), (2), (4), or (5) is also qualified as a general supervisor; therefore, depending upon the size and functions of the laboratory, the laboratory director may also serve as the laboratory supervisor; or
(2)
(i) Is a physician or has earned a doctoral degree from an accredited institution with a major in one of the chemical, physical, or biological sciences; and
(ii) Subsequent to graduation, has had at least 2 years of experience in one of the laboratory specialties in a laboratory; or
(3)
(i) Holds a master's degree from an accredited institution with a major in one of the chemical, physical, or biological sciences; and
(ii) Subsequent to graduation has had at least 4 years of pertinent full-time laboratory experience of which not less than 2 years have been spent working in the designated specialty in a laboratory; or
(4)
(i) Is qualified as a laboratory technologist under § 493.1491; and
(ii) After qualifying as a laboratory technologist, has had at least 6 years of pertinent full-time laboratory experience of which not less than 2 years have been spent working in the designated laboratory specialty in a laboratory; or
(5) With respect to individuals first qualifying before July 1, 1971, has had at least 15 years of pertinent full-time laboratory experience before January 1, 1968; this required experience may be met by the substitution of education for experience.
[58 FR 39155, July 22, 1993]
§ 493.1463 Standard: General supervisor responsibilities.
The general supervisor is responsible for day-to-day supervision or oversight of the laboratory operation and personnel performing testing and reporting test results.
(a) The general supervisor—
(1) Must be accessible to testing personnel at all times testing is performed to provide on-site, telephone or electronic consultation to resolve technical problems in accordance with policies and procedures established either by the laboratory director or technical supervisor;
(2) Is responsible for providing day-to-day supervision of high complexity test performance by a testing personnel qualified under § 493.1489;
(3) Except as specified in paragraph (c) of this section, must be onsite to provide direct supervision when high complexity testing is performed by any individuals qualified under § 493.1489(b)(5); and
(4) Is responsible for monitoring test analyses and specimen examinations to ensure that acceptable levels of analytic performance are maintained.
(b) The director or technical supervisor may delegate to the general supervisor the responsibility for—
(1) Assuring that all remedial actions are taken whenever test systems deviate from the laboratory's established performance specifications;
(2) Ensuring that patient test results are not reported until all corrective actions have been taken and the test system is properly functioning;
(3) Providing orientation to all testing personnel; and
(4) Annually evaluating and documenting the performance of all testing personnel.
(c) Exception. For individuals qualified under § 493.1489(b)(5), who were performing high complexity testing on or before January 19, 1993, the requirements of paragraph (a)(3) of this section are not effective, provided that all high complexity testing performed by the individual in the absence of a general supervisor is reviewed within 24 hours by a general supervisor qualified under § 493.1461.
[57 FR 7172, Feb. 28, 1992, as amended at 58 FR 5235, Jan. 19, 1993; 60 FR 20050, Apr. 24, 1995]
§ 493.1467 Condition: Laboratories performing high complexity testing; cytology general supervisor.
For the subspecialty of cytology, the laboratory must have a general supervisor who meets the qualification requirements of § 493.1469 of this subpart, and provides supervision in accordance with § 493.1471 of this subpart.
§ 493.1469 Standard: Cytology general supervisor qualifications.
The cytology general supervisor must be qualified to supervise cytology services. The general supervisor in cytology must possess a current license issued by the State in which the laboratory is located, if such licensing is required, and must—
(a) Be qualified as a technical supervisor under § 493.1449 (b) or (k); or
(b)
(1) Be qualified as a cytotechnologist under § 493.1483; and
(2) Have at least 3 years of full-time (2,080 hours per year) experience as a cytotechnologist within the preceding 10 years.
§ 493.1471 Standard: Cytology general supervisor responsibilities.
The technical supervisor of cytology may perform the duties of the cytology general supervisor or delegate the responsibilities to an individual qualified under § 493.1469.
(a) The cytology general supervisor is responsible for the day-to-day supervision or oversight of the laboratory operation and personnel performing testing and reporting test results.
(b) The cytology general supervisor must—
(1) Be accessible to provide on-site, telephone, or electronic consultation to resolve technical problems in accordance with policies and procedures established by the technical supervisor of cytology;
(2) Document the slide interpretation results of each gynecologic and nongynecologic cytology case he or she examined or reviewed (as specified under § 493.1274(c));
(3) For each 24-hour period, document the total number of slides he or she examined or reviewed in the laboratory as well as the total number of slides examined or reviewed in any other laboratory or for any other employer; and
(4) Document the number of hours spent examining slides in each 24-hour period.
[57 FR 7172, Feb. 28, 1992, as amended at 68 FR 3714, Jan. 24, 2003]
§ 493.1481 Condition: Laboratories performing high complexity testing; cytotechnologist.
For the subspecialty of cytology, the laboratory must have a sufficient number of cytotechnologists who meet the qualifications specified in § 493.1483 to perform the functions specified in § 493.1485.
§ 493.1483 Standard: Cytotechnologist qualifications.
Each person examining cytology slide preparations must meet the qualifications of § 493.1449 (b) or (k), or—
(a) Possess a current license as a cytotechnologist issued by the State in which the laboratory is located, if such licensing is required; and
(b) Meet one of the following requirements:
(1) Have graduated from a school of cytotechnology accredited by the Committee on Allied Health Education and Accreditation or other organization approved by HHS; or
(2) Be certified in cytotechnology by a certifying agency approved by HHS; or
(3) Before September 1, 1992—
(i) Have successfully completed 2 years in an accredited institution with at least 12 semester hours in science, 8 hours of which are in biology; and
(A) Have had 12 months of training in a school of cytotechnology accredited by an accrediting agency approved by HHS; or
(B) Have received 6 months of formal training in a school of cytotechnology accredited by an accrediting agency approved by HHS and 6 months of full-time experience in cytotechnology in a laboratory acceptable to the pathologist who directed the formal 6 months of training; or
(ii) Have achieved a satisfactory grade to qualify as a cytotechnologist in a proficiency examination approved by HHS and designed to qualify persons as cytotechnologists; or
(4) Before September 1, 1994, have full-time experience of at least 2 years or equivalent within the preceding 5 years examining slide preparations under the supervision of a physician qualified under § 493.1449(b) or (k)(1), and before January 1, 1969, must have—
(i) Graduated from high school;
(ii) Completed 6 months of training in cytotechnology in a laboratory directed by a pathologist or other physician providing cytology services; and
(iii) Completed 2 years of full-time supervised experience in cytotechnology; or
(5)
(i) On or before September 1, 1994, have full-time experience of at least 2 years or equivalent examining cytology slide preparations within the preceding 5 years in the United States under the supervision of a physician qualified under § 493.1449(b) or (k)(1); and
(ii) On or before September 1, 1995, have met the requirements in either paragraph (b)(1) or (2) of this section.
[57 FR 7172, Feb. 28, 1992, as amended at 59 FR 685, Jan. 6, 1994]
§ 493.1485 Standard; Cytotechnologist responsibilities.
The cytotechnologist is responsible for documenting—
(a) The slide interpretation results of each gynecologic and nongynecologic cytology case he or she examined or reviewed (as specified in § 493.1274(c));
(b) For each 24-hour period, the total number of slides examined or reviewed in the laboratory as well as the total number of slides examined or reviewed in any other laboratory or for any other employer; and
(c) The number of hours spent examining slides in each 24-hour period.
[57 FR 7172, Feb. 28, 1992, as amended at 68 FR 3714, Jan. 24, 2003]
§ 493.1487 Condition: Laboratories performing high complexity testing; testing personnel.
The laboratory has a sufficient number of individuals who meet the qualification requirements of § 493.1489 of this subpart to perform the functions specified in § 493.1495 of this subpart for the volume and complexity of testing performed.
§ 493.1489 Standard; Testing personnel qualifications.
Each individual performing high complexity testing must—
(a) Possess a current license issued by the State in which the laboratory is located, if such licensing is required; and
(b) Meet one of the following requirements:
(1) Be a doctor of medicine, doctor of osteopathy, or doctor of podiatric medicine licensed to practice medicine, osteopathy, or podiatry in the State in which the laboratory is located or have earned a doctoral, master's or bachelor's degree in a chemical, physical, biological or clinical laboratory science, or medical technology from an accredited institution;
(2)
(i) Have earned an associate degree in a laboratory science, or medical laboratory technology from an accredited institution or—
(ii) Have education and training equivalent to that specified in paragraph (b)(2)(i) of this section that includes—
(A) At least 60 semester hours, or equivalent, from an accredited institution that, at a minimum, include either—
(1) 24 semester hours of medical laboratory technology courses; or
(2) 24 semester hours of science courses that include—
(i) Six semester hours of chemistry;
(ii) Six semester hours of biology; and
(iii) Twelve semester hours of chemistry, biology, or medical laboratory technology in any combination; and
(B) Have laboratory training that includes either of the following:
(1) Completion of a clinical laboratory training program approved or accredited by the ABHES, the CAHEA, or other organization approved by HHS. (This training may be included in the 60 semester hours listed in paragraph (b)(2)(ii)(A) of this section.)
(2) At least 3 months documented laboratory training in each specialty in which the individual performs high complexity testing.
(3) Have previously qualified or could have qualified as a technologist under § 493.1491 on or before February 28, 1992;
(4) On or before April 24, 1995 be a high school graduate or equivalent and have either—
(i) Graduated from a medical laboratory or clinical laboratory training program approved or accredited by ABHES, CAHEA, or other organization approved by HHS; or
(ii) Successfully completed an official U.S. military medical laboratory procedures training course of at least 50 weeks duration and have held the military enlisted occupational specialty of Medical Laboratory Specialist (Laboratory Technician);
(5)
(i) Until September 1, 1997—
(A) Have earned a high school diploma or equivalent; and
(B) Have documentation of training appropriate for the testing performed before analyzing patient specimens. Such training must ensure that the individual has—
(1) The skills required for proper specimen collection, including patient preparation, if applicable, labeling, handling, preservation or fixation, processing or preparation, transportation and storage of specimens;
(2) The skills required for implementing all standard laboratory procedures;
(3) The skills required for performing each test method and for proper instrument use;
(4) The skills required for performing preventive maintenance, troubleshooting, and calibration procedures related to each test performed;
(5) A working knowledge of reagent stability and storage;
(6) The skills required to implement the quality control policies and procedures of the laboratory;
(7) An awareness of the factors that influence test results; and
(8) The skills required to assess and verify the validity of patient test results through the evaluation of quality control values before reporting patient test results; and
(ii) As of September 1, 1997, be qualified under § 493.1489(b)(1), (b)(2), or (b)(4), except for those individuals qualified under paragraph (b)(5)(i) of this section who were performing high complexity testing on or before April 24, 1995;
(6) For blood gas analysis—
(i) Be qualified under § 493.1489(b)(1), (b)(2), (b)(3), (b)(4), or (b)(5);
(ii) Have earned a bachelor's degree in respiratory therapy or cardiovascular technology from an accredited institution; or
(iii) Have earned an associate degree related to pulmonary function from an accredited institution; or
(7) For histopathology, meet the qualifications of § 493.1449 (b) or (l) to perform tissue examinations.
[57 FR 7172, Feb. 28, 1992, as amended at 58 FR 5236, Jan. 19, 1993; 58 FR 39155, July 22, 1993; 60 FR 20050, Apr. 24, 1995]
§ 493.1491 Technologist qualifications on or before February 28, 1992.
In order to qualify as high complexity testing personnel under § 493.1489(b)(3), the individual must have met or could have met the following qualifications for technologist as they were in effect on or before February 28, 1992. Each technologist must—
(a) Possess a current license as a laboratory technologist issued by the State, if such licensing exists; and
(b)
(1) Have earned a bachelor's degree in medical technology from an accredited university; or
(2) Have successfully completed 3 years of academic study (a minimum of 90 semester hours or equivalent) in an accredited college or university, which met the specific requirements for entrance into a school of medical technology accredited by an accrediting agency approved by the Secretary, and has successfully completed a course of training of at least 12 months in such a school; or
(3) Have earned a bachelor's degree in one of the chemical, physical, or biological sciences and, in addition, has at least 1 year of pertinent full-time laboratory experience or training, or both, in the specialty or subspecialty in which the individual performs tests; or
(4)
(i) Have successfully completed 3 years (90 semester hours or equivalent) in an accredited college or university with the following distribution of courses—
(A) For those whose training was completed before September 15, 1963. At least 24 semester hours in chemistry and biology courses of which—
(1) At least 6 semester hours were in inorganic chemistry and at least 3 semester hours were in other chemistry courses; and
(2) At least 12 semester hours in biology courses pertinent to the medical sciences; or
(B) For those whose training was completed after September 14, 1963. (1) 16 semester hours in chemistry courses that included at least 6 semester hours in inorganic chemistry and that are acceptable toward a major in chemistry;
(2) 16 semester hours in biology courses that are pertinent to the medical sciences and are acceptable toward a major in the biological sciences; and
(3) 3 semester hours of mathematics; and
(ii) Has experience, training, or both, covering several fields of medical laboratory work of at least 1 year and of such quality as to provide him or her with education and training in medical technology equivalent to that described in paragraphs (b)(1) and (2) of this section; or
(5) With respect to individuals first qualifying before July 1, 1971, the technologist—
(i) Was performing the duties of a laboratory technologist at any time between July 1, 1961, and January 1, 1968, and
(ii) Has had at least 10 years of pertinent laboratory experience prior to January 1, 1968. (This required experience may be met by the substitution of education for experience); or
(6) Achieves a satisfactory grade in a proficiency examination approved by HHS.
[58 FR 39155, July 22, 1993]
§ 493.1495 Standard; Testing personnel responsibilities.
The testing personnel are responsible for specimen processing, test performance and for reporting test results.
(a) Each individual performs only those high complexity tests that are authorized by the laboratory director and require a degree of skill commensurate with the individual's education, training or experience, and technical abilities.
(b) Each individual performing high complexity testing must—
(1) Follow the laboratory's procedures for specimen handling and processing, test analyses, reporting and maintaining records of patient test results;
(2) Maintain records that demonstrate that proficiency testing samples are tested in the same manner as patient specimens;
(3) Adhere to the laboratory's quality control policies, document all quality control activities, instrument and procedural calibrations and maintenance performed;
(4) Follow the laboratory's established policies and procedures whenever test systems are not within the laboratory's established acceptable levels of performance;
(5) Be capable of identifying problems that may adversely affect test performance or reporting of test results and either must correct the problems or immediately notify the general supervisor, technical supervisor, clinical consultant, or director;
(6) Document all corrective actions taken when test systems deviate from the laboratory's established performance specifications; and
(7) Except as specified in paragraph (c) of this section, if qualified under § 493.1489(b)(5), perform high complexity testing only under the onsite, direct supervision of a general supervisor qualified under § 493.1461.
(c) Exception. For individuals qualified under § 493.1489(b)(5), who were performing high complexity testing on or before January 19, 1993, the requirements of paragraph (b)(7) of this section are not effective, provided that all high complexity testing performed by the individual in the absence of a general supervisor is reviewed within 24 hours by a general supervisor qualified under § 493.1461.
[57 FR 7172, Feb. 28, 1992, as amended at 58 FR 5236, Jan. 19, 1993; 60 FR 20050, Apr. 24, 1995]
Subparts N-P [Reserved]
Subpart Q - Inspection
Source:
57 FR 7184, Feb. 28, 1992, unless otherwise noted.
§ 493.1771 Condition: Inspection requirements applicable to all CLIA-certified and CLIA-exempt laboratories.
(a) Each laboratory issued a CLIA certificate must meet the requirements in § 493.1773 and the specific requirements for its certificate type, as specified in §§ 493.1775 through 493.1780.
(b) All CLIA-exempt laboratories must comply with the inspection requirements in §§ 493.1773 and 493.1780, when applicable.
[63 FR 26737, May 14, 1998]
§ 493.1773 Standard: Basic inspection requirements for all laboratories issued a CLIA certificate and CLIA-exempt laboratories.
(a) A laboratory issued a certificate must permit CMS or a CMS agent to conduct an inspection to assess the laboratory's compliance with the requirements of this part. A CLIA-exempt laboratory and a laboratory that requests, or is issued a certificate of accreditation, must permit CMS or a CMS agent to conduct validation and complaint inspections.
(b) General requirements. As part of the inspection process, CMS or a CMS agent may require the laboratory to do the following:
(1) Test samples, including proficiency testing samples, or perform procedures.
(2) Permit interviews of all personnel concerning the laboratory's compliance with the applicable requirements of this part.
(3) Permit laboratory personnel to be observed performing all phases of the total testing process (preanalytic, analytic, and postanalytic).
(4) Permit CMS or a CMS agent access to all areas encompassed under the certificate including, but not limited to, the following:
(i) Specimen procurement and processing areas.
(ii) Storage facilities for specimens, reagents, supplies, records, and reports.
(iii) Testing and reporting areas.
(5) Provide CMS or a CMS agent with copies or exact duplicates of all records and data it requires.
(c) Accessible records and data. A laboratory must have all records and data accessible and retrievable within a reasonable time frame during the course of the inspection.
(d) Requirement to provide information and data. A laboratory must provide, upon request, all information and data needed by CMS or a CMS agent to make a determination of the laboratory's compliance with the applicable requirements of this part.
(e) Reinspection. CMS or a CMS agent may reinspect a laboratory at any time to evaluate the ability of the laboratory to provide accurate and reliable test results.
(f) Complaint inspection. CMS or a CMS agent may conduct an inspection when there are complaints alleging noncompliance with any of the requirements of this part.
(g) Failure to permit an inspection or reinspection. Failure to permit CMS or a CMS agent to conduct an inspection or reinspection results in the suspension or cancellation of the laboratory's participation in Medicare and Medicaid for payment, and suspension or limitation of, or action to revoke the laboratory's CLIA certificate, in accordance with subpart R of this part.
[63 FR 26737, May 14, 1998; 63 FR 32699, June 15, 1998]
§ 493.1775 Standard: Inspection of laboratories issued a certificate of waiver or a certificate for provider-performed microscopy procedures.
(a) A laboratory that has been issued a certificate of waiver or a certificate for provider-performed microscopy procedures is not subject to biennial inspections.
(b) If necessary, CMS or a CMS agent may conduct an inspection of a laboratory issued a certificate of waiver or a certificate for provider-performed microscopy procedures at any time during the laboratory's hours of operation to do the following:
(1) Determine if the laboratory is operated and testing is performed in a manner that does not constitute an imminent and serious risk to public health.
(2) Evaluate a complaint from the public.
(3) Determine whether the laboratory is performing tests beyond the scope of the certificate held by the laboratory.
(4) Collect information regarding the appropriateness of tests specified as waived tests or provider-performed microscopy procedures.
(c) The laboratory must comply with the basic inspection requirements of § 493.1773.
[63 FR 26737, May 14, 1998]
§ 493.1777 Standard: Inspection of laboratories that have requested or have been issued a certificate of compliance.
(a) Initial inspection.
(1) A laboratory issued a registration certificate must permit an initial inspection to assess the laboratory's compliance with the requirements of this part before CMS issues a certificate of compliance.
(2) The inspection may occur at any time during the laboratory's hours of operation.
(b) Subsequent inspections.
(1) CMS or a CMS agent may conduct subsequent inspections on a biennial basis or with such other frequency as CMS determines to be necessary to ensure compliance with the requirements of this part.
(2) CMS bases the nature of subsequent inspections on the laboratory's compliance history.
(c) Provider-performed microscopy procedures. The inspection sample for review may include testing in the subcategory of provider-performed microscopy procedures.
(d) Compliance with basic inspection requirements. The laboratory must comply with the basic inspection requirements of § 493.1773.
[63 FR 26738, May 14, 1998]
§ 493.1780 Standard: Inspection of CLIA-exempt laboratories or laboratories requesting or issued a certificate of accreditation.
(a) Validation inspection. CMS or a CMS agent may conduct a validation inspection of any accredited or CLIA-exempt laboratory at any time during its hours of operation.
(b) Complaint inspection. CMS or a CMS agent may conduct a complaint inspection of a CLIA-exempt laboratory or a laboratory requesting or issued a certificate of accreditation at any time during its hours of operation upon receiving a complaint applicable to the requirements of this part.
(c) Noncompliance determination. If a validation or complaint inspection results in a finding that the laboratory is not in compliance with one or more condition-level requirements, the following actions occur:
(1) A laboratory issued a certificate of accreditation is subject to a full review by CMS, in accordance with subpart E of this part and § 488.11 of this chapter.
(2) A CLIA-exempt laboratory is subject to appropriate enforcement actions under the approved State licensure program.
(d) Compliance with basic inspection requirements. CLIA-exempt laboratories and laboratories requesting or issued a certificate of accreditation must comply with the basic inspection requirements in § 493.1773.
[63 FR 26738, May 14, 1998]
Subpart R - Enforcement Procedures
Source:
57 FR 7237, Feb. 28, 1992, unless otherwise noted.
§ 493.1800 Basis and scope.
(a) Statutory basis.
(1) Section 1846 of the Act—
(i) Provides for intermediate sanctions that may be imposed on laboratories that perform clinical diagnostic tests on human specimens when those laboratories are found to be out of compliance with one or more of the conditions for Medicare coverage of their services; and
(ii) Requires the Secretary to develop and implement a range of such sanctions, including four that are specified in the statute.
(2) The Clinical Laboratory Improvement Act of 1967 (section 353 of the Public Health Service Act) as amended by CLIA 1988, as amended by section 2 of the Taking Essential Steps for Testing Act of 2012—
(i) Establishes requirements for all laboratories that perform clinical diagnostic tests on human specimens;
(ii) Requires a Federal certification scheme to be applied to all such laboratories; and
(iii) Grants the Secretary broad enforcement authority, including—
(A) Use of intermediate sanctions;
(B) Suspension, limitation, or revocation of the certificate of a laboratory that is out of compliance with one or more requirements for a certificate; and
(C) Civil suit to enjoin any laboratory activity that constitutes a significant hazard to the public health.
(3) Section 353 also—
(i) Provides for imprisonment or fine for any person convicted of intentional violation of CLIA requirements;
(ii) Specifies the administrative hearing and judicial review rights of a laboratory that is sanctioned under CLIA; and
(iii) Requires the Secretary to publish annually a list of all laboratories that have been sanctioned during the preceding year.
(b) Scope and applicability. This subpart sets forth—
(1) The policies and procedures that CMS follows to enforce the requirements applicable to laboratories under CLIA and under section 1846 of the Act; and
(2) The appeal rights of laboratories on which CMS imposes sanctions.
[57 FR 7237, Feb. 28, 1992, as amended at 79 FR 25480, May 2, 2014]
§ 493.1804 General considerations.
(a) Purpose. The enforcement mechanisms set forth in this subpart have the following purposes:
(1) To protect all individuals served by laboratories against substandard testing of specimens.
(2) To safeguard the general public against health and safety hazards that might result from laboratory activities.
(3) To motivate laboratories to comply with CLIA requirements so that they can provide accurate and reliable test results.
(b) Basis for decision to impose sanctions.
(1) CMS's decision to impose sanctions is based on one or more of the following:
(i) Deficiencies found by CMS or its agents in the conduct of inspections to certify or validate compliance with Federal requirements, or through review of materials submitted by the laboratory (e.g., personnel qualifications).
(ii) Unsuccessful participation in proficiency testing.
(2) CMS imposes one or more of the alternative or principal sanctions specified in §§ 493.1806 and 493.1807 when CMS or CMS's agent finds that a laboratory has condition-level deficiencies.
(c) Imposition of alternative sanctions.
(1) CMS may impose alternative sanctions in lieu of, or in addition to principal sanctions. (Except for a condition level deficiency under §§ 493.41 or 493.1100(a), CMS does not impose alternative sanctions on laboratories that have certificates of waiver because those laboratories are not routinely inspected for compliance with condition-level requirements.)
(2) CMS may impose alternative sanctions other than a civil money penalty after the laboratory has had an opportunity to respond, but before the hearing specified in § 493.1844.
(d) Choice of sanction: Factors considered. CMS bases its choice of sanction or sanctions on consideration of one or more factors that include, but are not limited to, the following, as assessed by the State or by CMS, or its agents:
(1) Whether the deficiencies pose immediate jeopardy.
(2) The nature, incidence, severity, and duration of the deficiencies or noncompliance.
(3) Whether the same condition level deficiencies have been identified repeatedly.
(4) The accuracy and extent of laboratory records (e.g., of remedial action) in regard to the noncompliance, and their availability to the State, to other CMS agents, and to CMS.
(5) The relationship of one deficiency or group of deficiencies to other deficiencies.
(6) The overall compliance history of the laboratory including but not limited to any period of noncompliance that occurred between certifications of compliance.
(7) The corrective and long-term compliance outcomes that CMS hopes to achieve through application of the sanction.
(8) Whether the laboratory has made any progress toward improvement following a reasonable opportunity to correct deficiencies.
(9) Any recommendation by the State agency as to which sanction would be appropriate.
(e) Number of alternative sanctions. CMS may impose a separate sanction for each condition level deficiency or a single sanction for all condition level deficiencies that are interrelated and subject to correction by a single course of action.
(f) Appeal rights. The appeal rights of laboratories dissatisfied with the imposition of a sanction are set forth in § 493.1844.
[57 FR 7237, Feb. 28, 1992; 57 FR 35761, Aug. 11, 1992, as amended at 60 FR 20051, Apr. 24, 1995; 85 FR 54874, Sept. 2, 2020]
§ 493.1806 Available sanctions: All laboratories.
(a) Applicability. CMS may impose one or more of the sanctions specified in this section on a laboratory that is out of compliance with one or more CLIA conditions.
(b) Principal sanction. CMS may impose any of the three principal CLIA sanctions, which are suspension, limitation, or revocation of any type of CLIA certificate.
(c) Alternative sanctions. CMS may impose one or more of the following alternative sanctions in lieu of or in addition to imposing a principal sanction, except on a laboratory that has a certificate of waiver.
(1) Directed plan of correction, as set forth at § 493.1832.
(2) State onsite monitoring as set forth at § 493.1836.
(3) Civil money penalty, as set forth at § 493.1834.
(d) Civil suit. CMS may bring suit in the appropriate U.S. District Court to enjoin continuation of any activity of any laboratory (including a CLIA-exempt laboratory that has been found with deficiencies during a validation survey), if CMS has reason to believe that continuation of the activity would constitute a significant hazard to the public health.
(e) Criminal sanctions. Under section 353(1) of the PHS Act, an individual who is convicted of intentionally violating any CLIA requirement may be imprisoned or fined.
[57 FR 7237, Feb. 28, 1992, as amended at 58 FR 5237, Jan. 19, 1993]
§ 493.1807 Additional sanctions: Laboratories that participate in Medicare.
The following additional sanctions are available for laboratories that are out of compliance with one or more CLIA conditions and that have approval to receive Medicare payment for their services.
(a) Principal sanction. Cancellation of the laboratory's approval to receive Medicare payment for its services.
(b) Alternative sanctions.
(1) Suspension of payment for tests in one or more specific specialties or subspecialties, performed on or after the effective date of sanction.
(2) Suspension of payment for all tests in all specialties and subspecialties performed on or after the effective date of sanction.
§ 493.1808 Adverse action on any type of CLIA certificate: Effect on Medicare approval.
(a) Suspension or revocation of any type of CLIA certificate. When CMS suspends or revokes any type of CLIA certificate, CMS concurrently cancels the laboratory's approval to receive Medicare payment for its services.
(b) Limitation of any type of CLIA certificate. When CMS limits any type of CLIA certificate, CMS concurrently limits Medicare approval to only those specialties or subspecialties that are authorized by the laboratory's limited certificate.
§ 493.1809 Limitation on Medicaid payment.
As provided in section 1902(a)(9)(C) of the Act, payment for laboratory services may be made under the State plan only if those services are furnished by a laboratory that has a CLIA certificate or is licensed by a State whose licensure program has been approved by the Secretary under this part.
[57 FR 7237, Feb. 28, 1992; 57 FR 35761, Aug. 11, 1992]
§ 493.1810 Imposition and lifting of alternative sanctions.
(a) Notice of noncompliance and of proposed sanction: Content. If CMS or its agency identifies condition level noncompliance in a laboratory, CMS or its agent gives the laboratory written notice of the following:
(1) The condition level noncompliance that it has identified.
(2) The sanction or sanctions that CMS or its agent proposes to impose against the laboratory.
(3) The rationale for the proposed sanction or sanctions.
(4) The projected effective date and duration of the proposed sanction or sanctions.
(5) The authority for the proposed sanction or sanctions.
(6) The time allowed (at least 10 days) for the laboratory to respond to the notice.
(b) Opportunity to respond. During the period specified in paragraph (a)(6) of this section, the laboratory may submit to CMS or its agent written evidence or other information against the imposition of the proposed sanction or sanctions.
(c) Notice of imposition of sanction —
(1) Content. CMS gives the laboratory written notice that acknowledges any evidence or information received from the laboratory and specifies the following:
(i) The sanction or sanctions to be imposed against the laboratory.
(ii) The authority and rationale for the imposing sanction or sanctions.
(iii) The effective date and duration of sanction.
(2) Timing.
(i) If CMS or its agent determines that the deficiencies pose immediate jeopardy, CMS provides notice at least 5 days before the effective date of sanction.
(ii) If CMS or its agent determines that the deficiencies do not pose immediate jeopardy, CMS provides notice at least 15 days before the effective date of the sanction.
(d) Duration of alternative sanctions. An alternative sanction continues until the earlier of the following occurs:
(1) The laboratory corrects all condition level deficiencies.
(2) CMS's suspension, limitation, or revocation of the laboratory's CLIA certificate becomes effective.
(e) Lifting of alternative sanctions —
(1) General rule. Alternative sanctions are not lifted until a laboratory's compliance with all condition level requirements is verified.
(2) Credible allegation of compliance. When a sanctioned laboratory submits a credible allegation of compliance, CMS's agent determines whether—
(i) It can certify compliance on the basis of the evidence presented by the laboratory in its allegation; or
(ii) It must revisit to verify whether the laboratory has, in fact, achieved compliance.
(3) Compliance achieved before the date of revisit. If during a revisit, the laboratory presents credible evidence (as determined by CMS or its agent) that it achieved compliance before the date of revisit, sanctions are lifted as of that earlier date.
§ 493.1812 Action when deficiencies pose immediate jeopardy.
If a laboratory's deficiencies pose immediate jeopardy, the following rules apply:
(a) CMS requires the laboratory to take immediate action to remove the jeopardy and may impose one or more alternative sanctions to help bring the laboratory into compliance.
(b) If the findings of a revisit indicate that a laboratory has not eliminated the jeopardy, CMS suspends or limits the laboratory's CLIA certificate no earlier than 5 days after the date of notice of suspension or limitation. CMS may later revoke the certificate.
(c) In addition, if CMS has reason to believe that the continuation of any activity by any laboratory (either the entire laboratory operation or any specialty or subspecialty of testing) would constitute a significant hazard to the public health, CMS may bring suit and seek a temporary injunction or restraining order against continuation of that activity by the laboratory, regardless of the type of CLIA certificate the laboratory has and of whether it is State-exempt.
§ 493.1814 Action when deficiencies are at the condition level but do not pose immediate jeopardy.
If a laboratory has condition level deficiencies that do not pose immediate jeopardy, the following rules apply:
(a) Initial action.
(1) CMS may cancel the laboratory's approval to receive Medicare payment for its services.
(2) CMS may suspend, limit, or revoke the laboratory's CLIA certificate.
(3) If CMS does not impose a principal sanction under paragraph (a)(1) or (a)(2) of this section, it imposes one or more alternative sanctions. In the case of unsuccessful participation in proficiency testing, CMS may impose the training and technical assistance requirement set forth at § 493.1838 in lieu of, or in addition to, one or more alternative sanctions.
(b) Failure to correct condition level deficiencies. If CMS imposes alternative sanctions for condition level deficiencies that do not pose immediate jeopardy, and the laboratory does not correct the condition level deficiencies within 12 months after the last day of inspection, CMS—
(1) Cancels the laboratory's approval to receive Medicare payment for its services, and discontinues the Medicare payment sanctions as of the day cancellation is effective.
(2) Following a revisit which indicates that the laboratory has not corrected its condition level deficiencies, notifies the laboratory that it proposes to suspend, limit, or revoke the certificate, as specified in § 493.1816(b), and the laboratory's right to hearing; and
(3) May impose (or continue, if already imposed) any alternative sanctions that do not pertain to Medicare payments. (Sanctions imposed under the authority of section 353 of the PHS Act may continue for more than 12 months from the last date of inspection, while a hearing on the proposed suspension, limitation, or revocation of the certificate of compliance, registration certificate, certificate of accreditation, or certificate for PPM procedures is pending.)
(c) Action after hearing. If a hearing decision upholds a proposed suspension, limitation, or revocation of a laboratory's CLIA certificate, CMS discontinues any alternative sanctions as of the day it makes the suspension, limitation, or revocation effective.
[57 FR 7237, Feb. 28, 1992, as amended at 60 FR 20051, Apr. 24, 1995]
§ 493.1816 Action when deficiencies are not at the condition level.
If a laboratory has deficiencies, that are not at the condition level, the following rules apply:
(a) Initial action. The laboratory must submit a plan of correction that is acceptable to CMS in content and time frames.
(b) Failure to correct deficiencies. If, on revisit, it is found that the laboratory has not corrected the deficiencies within 12 months after the last day of inspection, the following rules apply:
(1) CMS cancels the laboratory's approval to receive Medicare payment for its services.
(2) CMS notifies the laboratory of its intent to suspend, limit, or revoke the laboratory's CLIA certificate and of the laboratory's right to a hearing.
§ 493.1820 Ensuring timely correction of deficiencies.
(a) Timing of visits. CMS, the State survey agency or other CMS agent may visit the laboratory at any time to evaluate progress, and at the end of the period to determine whether all corrections have been made.
(b) Deficiencies corrected before a visit. If during a visit, a laboratory produces credible evidence that it achieved compliance before the visit, the sanctions are lifted as of that earlier date.
(c) Failure to correct deficiencies. If during a visit it is found that the laboratory has not corrected its deficiencies, CMS may propose to suspend, limit, or revoke the laboratory's CLIA certificate.
(d) Additional time for correcting lower level deficiencies not at the condition level. If at the end of the plan of correction period all condition level deficiencies have been corrected, and there are deficiencies, that are not at the condition level, CMS may request a revised plan of correction. The revised plan may not extend beyond 12 months from the last day of the inspection that originally identified the cited deficiencies.
(e) Persistence of deficiencies. If at the end of the period covered by the plan of correction, the laboratory still has deficiencies, the rules of §§ 493.1814 and 493.1816 apply.
§ 493.1826 Suspension of part of Medicare payments.
(a) Application.
(1) CMS may impose this sanction if a laboratory—
(i) Is found to have condition level deficiencies with respect to one or more specialties or subspecialties of tests; and
(ii) Agrees (in return for not having its Medicare approval cancelled immediately) not to charge Medicare beneficiaries or their private insurance carriers for the services for which Medicare payment is suspended.
(2) CMS suspends Medicare payment for those specialities or subspecialties of tests for which the laboratory is out of compliance with Federal requirements.
(b) Procedures. Before imposing this sanction, CMS provides notice of sanction and opportunity to respond in accordance with § 493.1810.
(c) Duration and effect of sanction. This sanction continues until the laboratory corrects the condition level deficiencies or CMS cancels the laboratory's approval to receive Medicare payment for its services, but in no event longer than 12 months.
(1) If the laboratory corrects all condition level deficiencies, CMS resumes Medicare payment effective for all services furnished on or after the date the deficiencies are corrected.
(2) [Reserved]
[57 FR 7237, Feb. 28, 1992; 57 FR 35761, Aug. 11, 1992]
§ 493.1828 Suspension of all Medicare payments.
(a) Application.
(1) CMS may suspend payment for all Medicare-approved laboratory services when the laboratory has condition level deficiencies.
(2) CMS suspends payment for all Medicare covered laboratory services when the following conditions are met:
(i) Either—
(A) The laboratory has not corrected its condition level deficiencies included in the plan of correction within 3 months from the last date of inspection; or
(B) The laboratory has been found to have the same condition level deficiencies during three consecutive inspections; and
(ii) The laboratory has chosen (in return for not having its Medicare approval immediately cancelled), to not charge Medicare beneficiaries or their private insurance carriers for services for which Medicare payment is suspended.
(3) CMS suspends payment for services furnished on and after the effective date of sanction.
(b) Procedures. Before imposing this sanction, CMS provides notice of sanction and opportunity to respond in accordance with § 493.1810.
(c) Duration and effect of sanction.
(1) Suspension of payment continues until all condition level deficiencies are corrected, but never beyond twelve months.
(2) If all the deficiencies are not corrected by the end of the 12 month period, CMS cancels the laboratory's approval to receive Medicare payment for its services.
§ 493.1832 Directed plan of correction and directed portion of a plan of correction.
(a) Application. CMS may impose a directed plan of correction as an alternative sanction for any laboratory that has condition level deficiencies. If CMS does not impose a directed plan of correction as an alternative sanction for a laboratory that has condition level deficiencies, it at least imposes a directed portion of a plan of correction when it imposes any of the following alternative sanctions:
(1) State onsite monitoring.
(2) Civil money penalty.
(3) Suspension of all or part of Medicare payments.
(b) Procedures —
(1) Directed plan of correction. When imposing this sanction, CMS—
(i) Gives the laboratory prior notice of the sanction and opportunity to respond in accordance with § 493.1810;
(ii) Directs the laboratory to take specific corrective action within specific time frames in order to achieve compliance; and
(iii) May direct the laboratory to submit the names of laboratory clients for notification purposes, as specified in paragraph (b)(3) of this section.
(2) Directed portion of a plan of correction. CMS may decide to notify clients of a sanctioned laboratory, because of the seriousness of the noncompliance (e.g., the existence of immediate jeopardy) or for other reasons. When imposing this sanction, CMS takes the following steps—
(i) Directs the laboratory to submit to CMS, the State survey agency, or other CMS agent, within 10 calendar days after the notice of the alternative sanction, a list of names and addresses of all physicians, providers, suppliers, and other clients who have used some or all of the services of the laboratory since the last certification inspection or within any other timeframe specified by CMS.
(ii) Within 30 calendar days of receipt of the information, may send to each laboratory client, via the State survey agency, a notice containing the name and address of the laboratory, the nature of the laboratory's noncompliance, and the kind and effective date of the alternative sanction.
(iii) Sends to each laboratory client, via the State survey agency, notice of the recission of an adverse action within 30 days of the rescission.
(3) Notice of imposition of a principal sanction following the imposition of an alternative sanction. If CMS imposes a principal sanction following the imposition of an alternative sanction, and for which CMS has already obtained a list of laboratory clients, CMS may use that list to notify the clients of the imposition of the principal sanction.
(c) Duration of a directed plan of correction. If CMS imposes a directed plan of correction, and on revisit it is found that the laboratory has not corrected the deficiencies within 12 months from the last day of inspection, the following rules apply:
(1) CMS cancels the laboratory's approval for Medicare payment of its services, and notifies the laboratory of CMS's intent to suspend, limit, or revoke the laboratory's CLIA certificate.
(2) The directed plan of correction continues in effect until the day suspension, limitation, or revocation of the laboratory's CLIA certificate.
§ 493.1834 Civil money penalty.
(a) Statutory basis. Sections 1846 of the Act and 353(h)(2)(B) of the PHS Act authorize the Secretary to impose civil money penalties on laboratories. Section 1846(b)(3) of the Act specifically provides that incrementally more severe fines may be imposed for repeated or uncorrected deficiencies.
(b) Scope. This section sets forth the procedures that CMS follows to impose a civil money penalty in lieu of, or in addition to, suspending, limiting, or revoking the certificate of compliance, registration certificate, certificate of accreditation, or certificate for PPM procedures of a laboratory that is found to have condition level deficiencies.
(c) Basis for imposing a civil money penalty. CMS may impose a civil money penalty against any laboratory determined to have condition level deficiencies regardless of whether those deficiencies pose immediate jeopardy.
(d) Amount of penalty —
(1) Factors considered. In determining the amount of the penalty, CMS takes into account the following factors:
(i) The nature, scope, severity, and duration of the noncompliance.
(ii) Whether the same condition level deficiencies have been identified during three consecutive inspections.
(iii) The laboratory's overall compliance history including but not limited to any period of noncompliance that occurred between certifications of compliance.
(iv) The laboratory's intent or reason for noncompliance.
(v) The accuracy and extent of laboratory records and their availability to CMS, the State survey agency, or other CMS agent.
(2) Range of penalty amount.
(i) For a condition level deficiency that poses immediate jeopardy, the range is $3,050-$10,000 as adjusted annually under 45 CFR part 102 per day of noncompliance or per violation.
(ii) For a condition level deficiency that does not pose immediate jeopardy, the range is $50-$3,000 as adjusted annually under 45 CFR part 102 per day of noncompliance or per violation.
(iii) For a condition level deficiency under §§ 493.41 or 493.1100(a), the penalty amount is $1,000 for the first day of noncompliance and $500 for each additional day of noncompliance.
(3) Decreased penalty amounts. If the immediate jeopardy is removed, but the deficiency continues, CMS shifts the penalty amount to the lower range.
(4) Increased penalty amounts. CMS may, before the hearing, propose to increase the penalty amount for a laboratory that has deficiencies which, after imposition of a lower level penalty amount, become sufficiently serious to pose immediate jeopardy.
(e) Procedures for imposition of civil money penalty —
(1) Notice of intent.
(i) CMS sends the laboratory written notice, of CMS's intent to impose a civil money penalty.
(ii) The notice includes the following information:
(A) The statutory basis for the penalty.
(B) The proposed daily or per violation amount of the penalty.
(C) The factors (as described in paragraph (d)(1) of this section) that CMS considered.
(D) The opportunity for responding to the notice in accordance with § 493.1810(c).
(E) A specific statement regarding the laboratory's appeal rights.
(2) Appeal rights.
(i) The laboratory has 60 days from the date of receipt of the notice of intent to impose a civil money penalty to request a hearing in accordance with § 493.1844(g).
(ii) If the laboratory requests a hearing, all other pertinent provisions of § 493.1844 apply.
(iii) If the laboratory does not request a hearing, CMS may reduce the proposed penalty amount by 35 percent.
(f) Accrual and duration of penalty —
(1) Accrual of penalty. The civil money penalty begins accruing as follows:
(i) 5 days after notice of intent if there is immediate jeopardy.
(ii) 15 days after notice of intent if there is not immediate jeopardy.
(2) Duration of penalty. The civil money penalty continues to accrue until the earliest of the following occurs:
(i) The laboratory's compliance with condition level requirements is verified on the basis of the evidence presented by the laboratory in its credible allegation of compliance or at the time or revisit.
(ii) Based on credible evidence presented by the laboratory at the time of revisit, CMS determines that compliance was achieved before the revisit. (In this situation, the money penalty stops accruing as of the date of compliance.)
(iii) CMS suspends, limits, or revokes the laboratory's certificate of compliance, registration certificate, certificate of accreditation, or certificate for PPM procedures.
(g) Computation and notice of total penalty amount —
(1) Computation. CMS computes the total penalty amount after the laboratory's compliance is verified or CMS suspends, limits, or revokes the laboratory's CLIA certificate but in no event before—
(i) The 60 day period for requesting a hearing has expired without a request or the laboratory has explicitly waived its right to a hearing; or
(ii) Following a hearing requested by the laboratory, the ALJ issues a decision that upholds imposition of the penalty.
(2) Notice of penalty amount and due date of penalty. The notice includes the following information:
(i) Daily or per violation penalty amount.
(ii) Number of days or violations for which the penalty is imposed.
(iii) Total penalty amount.
(iv) Due date for payment of the penalty.
(h) Due date for payment of penalty.
(1) Payment of a civil money penalty is due 15 days from the date of the notice specified in paragraph (g)(2) of this section.
(2) CMS may approve a plan for a laboratory to pay a civil money penalty, plus interest, over a period of up to one year from the original due date.
(i) Collection and settlement —
(1) Collection of penalty amounts.
(i) The determined penalty amount may be deducted from any sums then or later owing by the United States to the laboratory subject to the penalty.
(ii) Interest accrues on the unpaid balance of the penalty, beginning on the due date. Interest is computed at the rate specified in § 405.378(d) of this chapter.
(2) Settlement. CMS has authority to settle any case at any time before the ALJ issues a hearing decision.
[57 FR 7237, Feb. 28, 1992, as amended at 60 FR 20051, Apr. 24, 1995; 61 FR 63749, Dec. 2, 1996; 81 FR 61564, Sept. 6, 2016; 85 FR 54874, Sept. 2, 2020]
§ 493.1836 State onsite monitoring.
(a) Application.
(1) CMS may require continuous or intermittent monitoring of a plan of correction by the State survey agency to ensure that the laboratory makes the improvements necessary to bring it into compliance with the condition level requirements. (The State monitor does not have management authority, that is, cannot hire or fire staff, obligate funds, or otherwise dictate how the laboratory operates. The monitor's responsibility is to oversee whether corrections are made.)
(2) The laboratory must pay the costs of onsite monitoring by the State survey agency.
(i) The costs are computed by multiplying the number of hours of onsite monitoring in the laboratory by the hourly rate negotiated by CMS and the State.
(ii) The hourly rate includes salary, fringe benefits, travel, and other direct and indirect costs approved by CMS.
(b) Procedures. Before imposing this sanction, CMS provides notice of sanction and opportunity to respond in accordance with § 493.1810.
(c) Duration of sanction.
(1) If CMS imposes onsite monitoring, the sanction continues until CMS determines that the laboratory has the capability to ensure compliance with all condition level requirements.
(2) If the laboratory does not correct all deficiencies within 12 months, and a revisit indicates that deficiencies remain, CMS cancels the laboratory's approval for Medicare payment for its services and notifies the laboratory of its intent to suspend, limit, or revoke the laboratory's certificate of compliance, registration certificate, certificate of accreditation, or certificate for PPM procedures.
(3) If the laboratory still does not correct its deficiencies, the Medicare sanction continues until the suspension, limitation, or revocation of the laboratory's certificate of compliance, registration certificate, certificate of accreditation, or certificate for PPM procedures is effective.
[57 FR 7237, Feb. 28, 1992, as amended at 60 FR 20051, Apr. 24, 1995]
§ 493.1838 Training and technical assistance for unsuccessful participation in proficiency testing.
If a laboratory's participation in proficiency testing is unsuccessful, CMS may require the laboratory to undertake training of its personnel, or to obtain necessary technical assistance, or both, in order to meet the requirements of the proficiency testing program. This requirement is separate from the principal and alternative sanctions set forth in §§ 493.1806 and 493.1807.
§ 493.1840 Suspension, limitation, or revocation of any type of CLIA certificate.
(a) Adverse action based on actions of the laboratory's owner, operator or employees. CMS may initiate adverse action to suspend, limit or revoke any CLIA certificate if CMS finds that a laboratory's owner or operator or one of its employees has—
(1) Been guilty of misrepresentation in obtaining a CLIA certificate;
(2) Performed, or represented the laboratory as entitled to perform, a laboratory examination or other procedure that is not within a category of laboratory examinations or other procedures authorized by its CLIA certificate;
(3) Failed to comply with the certificate requirements and performance standards;
(4) Failed to comply with reasonable requests by CMS for any information or work on materials that CMS concludes is necessary to determine the laboratory's continued eligibility for its CLIA certificate or continued compliance with performance standards set by CMS;
(5) Refused a reasonable request by CMS or its agent for permission to inspect the laboratory and its operation and pertinent records during the hours that the laboratory is in operation;
(6) Violated or aided and abetted in the violation of any provisions of CLIA and its implementing regulations;
(7) Failed to comply with an alternative sanction imposed under this subpart; or
(8) Within the preceding two-year period, owned or operated a laboratory that had its CLIA certificate revoked. (This provision applies only to the owner or operator, not to all of the laboratory's employees.)
(b) Adverse action based on improper referrals in proficiency testing. If CMS determines that a laboratory has intentionally referred its proficiency testing samples to another laboratory for analysis, CMS does one of the following:
(1)
(i) Revokes the laboratory's CLIA certificate for at least 1 year, prohibits the owner and operator from owning or operating a CLIA-certified laboratory for at least 1 year, and may impose a civil money penalty in accordance with § 493.1834(d), if CMS determines that—
(A) A proficiency testing referral is a repeat proficiency testing referral as defined at § 493.2; or
(B) On or before the proficiency testing event close date, a laboratory reported proficiency testing results obtained from another laboratory to the proficiency testing program.
(ii) Following the revocation of a CLIA certificate in accordance with paragraph (b)(1)(i) of this section, CMS may exempt a laboratory owner from the generally applicable prohibition on owning or operating a CLIA-certified laboratory under paragraph (a)(8) of this section on a laboratory-by-laboratory basis if CMS finds, after review of the relevant facts and circumstances, that there is no evidence that—
(A) Patients would be put at risk as a result of the owner being exempted from the ban on a laboratory-by-laboratory basis;
(B) The laboratory for which the owner is to be exempted from the general ownership ban participated in or was otherwise complicit in the PT referral of the laboratory that resulted in the revocation; and
(C) The laboratory for which the owner is to be exempted from the general ownership ban received a PT sample from another laboratory in the prior two survey cycles, and failed to immediately report such receipt to CMS or to the appropriate CMS-approved accrediting organization.
(2) Suspends or limits the CLIA certificate for less than 1 year based on the criteria in § 493.1804(d) and imposes alternative sanctions as appropriate, in accordance with §§ 493.1804(c) and (d), 493.1806(c), 493.1807(b), 493.1809 and, in the case of civil money penalties, § 493.1834(d), when CMS determines that paragraph (b)(1)(i)(A) or (B) of this section does not apply but that the laboratory obtained test results for the proficiency testing samples from another laboratory on or before the proficiency testing event close date. Among other possibilities, alternative sanctions will always include a civil money penalty and a directed plan of correction that includes required training of staff.
(3) Imposes alternative sanctions in accordance with §§ 493.1804(c) and (d), 493.1806(c), 493.1807(b), 493.1809 and, in the case of civil money penalties, § 493.1834(d), when CMS determines that paragraph (b)(1)(i) or (2) of this section do not apply, and a PT referral has occurred, but no test results are received prior to the event close date by the referring laboratory from the laboratory that received the referral. Among other possibilities, alternative sanctions will always include a civil money penalty and a directed plan of correction that includes required training of staff.
(c) Adverse action based on exclusion from Medicare. If the OIG excludes a laboratory from participation in Medicare, CMS suspends the laboratory's CLIA certificate for the period during which the laboratory is excluded.
(d) Procedures for suspension or limitation —
(1) Basic rule. Except as provided in paragraph (d)(2) of this section, CMS does not suspend or limit a CLIA certificate until after an ALJ hearing decision (as provided in § 493.1844) that upholds suspension or limitation.
(2) Exceptions. CMS may suspend or limit a CLIA certificate before the ALJ hearing in any of the following circumstances:
(i) The laboratory's deficiencies pose immediate jeopardy.
(ii) The laboratory has refused a reasonable request for information or work on materials.
(iii) The laboratory has refused permission for CMS or a CMS agent to inspect the laboratory or its operation.
(e) Procedures for revocation.
(1) CMS does not revoke any type of CLIA certificate until after an ALJ hearing that upholds revocation.
(2) CMS may revoke a CLIA certificate after the hearing decision even if it had not previously suspended or limited that certificate.
(f) Notice to the OIG. CMS notifies the OIG of any violations under paragraphs (a)(1), (a)(2), (a)(6), and (b) of this section within 30 days of the determination of the violation.
[57 FR 7237, Feb. 28, 1992, as amended at 79 FR 25480, May 2, 2014]
§ 493.1842 Cancellation of Medicare approval.
(a) Basis for cancellation.
(1) CMS always cancels a laboratory's approval to receive Medicare payment for its services if CMS suspends or revokes the laboratory's CLIA certificate.
(2) CMS may cancel the laboratory's approval under any of the following circumstances:
(i) The laboratory is out of compliance with a condition level requirement.
(ii) The laboratory fails to submit a plan of correction satisfactory to CMS.
(iii) The laboratory fails to correct all its deficiencies within the time frames specified in the plan of correction.
(b) Notice and opportunity to respond. Before canceling a laboratory's approval to receive Medicare payment for its services, CMS gives the laboratory—
(1) Written notice of the rationale for, effective date, and effect of, cancellation;
(2) Opportunity to submit written evidence or other information against cancellation of the laboratory's approval.
This sanction may be imposed before the hearing that may be requested by a laboratory, in accordance with the appeals procedures set forth in § 493.1844.
(c) Effect of cancellation. Cancellation of Medicare approval terminates any Medicare payment sanctions regardless of the time frames originally specified.
§ 493.1844 Appeals procedures.
(a) General rules.
(1) The provisions of this section apply to all laboratories and prospective laboratories that are dissatisfied with any initial determination under paragraph (b) of this section.
(2) Hearings are conducted in accordance with procedures set forth in subpart D of part 498 of this chapter, except that the authority to conduct hearings and issue decisions may be exercised by ALJs assigned to, or detailed to, the Departmental Appeals Board.
(3) Any party dissatisfied with a hearing decision is entitled to request review of the decision as specified in subpart E of part 498 of this chapter, except that the authority to review the decision may be exercised by the Departmental Appeals Board.
(4) When more than one of the actions specified in paragraph (b) of this section are carried out concurrently, the laboratory has a right to only one hearing on all matters at issue.
(b) Actions that are initial determinations. The following actions are initial determinations and therefore are subject to appeal in accordance with this section:
(1) The suspension, limitation, or revocation of the laboratory's CLIA certificate by CMS because of noncompliance with CLIA requirements.
(2) The denial of a CLIA certificate.
(3) The imposition of alternative sanctions under this subpart (but not the determination as to which alternative sanction or sanctions to impose).
(4) The denial or cancellation of the laboratory's approval to receive Medicare payment for its services.
(c) Actions that are not initial determinations. Actions that are not listed in paragraph (b) of this section are not initial determinations and therefore are not subject to appeal under this section. They include, but are not necessarily limited to, the following:
(1) The finding that a laboratory accredited by a CMS-approved accreditation organization is no longer deemed to meet the conditions set forth in subparts H, J, K, M, and Q of this part. However, the suspension, limitation or revocation of a certificate of accreditation is an initial determination and is appealable.
(2) The finding that a laboratory determined to be in compliance with condition-level requirements but has deficiencies that are not at the condition level.
(3) The determination not to reinstate a suspended CLIA certificate because the reason for the suspension has not been removed or there is insufficient assurance that the reason will not recur.
(4) The determination as to which alternative sanction or sanctions to impose, including the amount of a civil money penalty to impose per day or per violation.
(5) The denial of approval for Medicare payment for the services of a laboratory that does not have in effect a valid CLIA certificate.
(6) The determination that a laboratory's deficiencies pose immediate jeopardy.
(7) The amount of the civil money penalty assessed per day or for each violation of Federal requirements.
(d) Effect of pending appeals —
(1) Alternative sanctions. The effective date of an alternative sanction (other than a civil money penalty) is not delayed because the laboratory has appealed and the hearing or the hearing decision is pending.
(2) Suspension, limitation, or revocation of a laboratory's CLIA certificate —
(i) General rule. Except as provided in paragraph (d)(2)(ii) of this section, suspension, limitation, or revocation of a CLIA certificate is not effective until after a hearing decision by an ALJ is issued.
(ii) Exceptions.
(A) If CMS determines that conditions at a laboratory pose immediate jeopardy, the effective date of the suspension or limitation of a CLIA certificate is not delayed because the laboratory has appealed and the hearing or the hearing decision is pending.
(B) CMS may suspend or limit a laboratory's CLIA certificate before an ALJ hearing or hearing decision if the laboratory has refused a reasonable request for information (including but not limited to billing information), or for work on materials, or has refused permission for CMS or a CMS agent to inspect the laboratory or its operation.
(3) Cancellation of Medicare approval. The effective date of the cancellation of a laboratory's approval to receive Medicare payment for its services is not delayed because the laboratory has appealed and the hearing or hearing decision is pending.
(4) Effect of ALJ decision.
(i) An ALJ decision is final unless, as provided in paragraph (a)(3) of this section, one of the parties requests review by the Departmental Appeals Board within 60 days, and the Board reviews the case and issues a revised decision.
(ii) If an ALJ decision upholds a suspension imposed because of immediate jeopardy, that suspension becomes a revocation.
(e) Appeal rights for prospective laboratories —
(1) Reconsideration. Any prospective laboratory dissatisfied with a denial of a CLIA certificate, or of approval for Medicare payment for its services, may initiate the appeals process by requesting reconsideration in accordance with §§ 498.22 through 498.25 of this chapter.
(2) Notice of reopening. If CMS reopens an initial or reconsidered determination, CMS gives the prospective laboratory notice of the revised determination in accordance with § 498.32 of this chapter.
(3) ALJ hearing. Any prospective laboratory dissatisfied with a reconsidered determination under paragraph (e)(1) of this section or a revised reconsidered determination under § 498.30 of this chapter is entitled to a hearing before an ALJ, as specified in paragraph (a)(2) of this section.
(4) Review of ALJ hearing decisions. Any prospective laboratory that is dissatisfied with an ALJ's hearing decision or dismissal of a request for hearing may file a written request for review by the Departmental Appeals Board as provided in paragraph (a)(3) of this section.
(f) Appeal rights of laboratories —
(1) ALJ hearing. Any laboratory dissatisfied with the suspension, limitation, or revocation of its CLIA certificate, with the imposition of an alternative sanction under this subpart, or with cancellation of the approval to receive Medicare payment for its services, is entitled to a hearing before an ALJ as specified in paragraph (a)(2) of this section and has 60 days from the notice of sanction to request a hearing.
(2) Review of ALJ hearing decisions. Any laboratory that is dissatisfied with an ALJ's hearing decision or dismissal of a request for hearing may file a written request for review by the Departmental Appeals Board, as provided in paragraph (a)(3) of this section.
(3) Judicial review. Any laboratory dissatisfied with the decision to impose a civil money penalty or to suspend, limit, or revoke its CLIA certificate may, within 60 days after the decision becomes final, file with the U.S. Court of Appeals of the circuit in which the laboratory has its principal place of business, a petition for judicial review.
(g) Notice of adverse action.
(1) If CMS suspends, limits, or revokes a laboratory's CLIA certificate or cancels the approval to receive Medicare payment for its services, CMS gives notice to the laboratory, and may give notice to physicians, providers, suppliers, and other laboratory clients, according to the procedures set forth at § 493.1832. In addition, CMS notifies the general public each time one of these principal sanctions is imposed.
(2) The notice to the laboratory—
(i) Sets forth the reasons for the adverse action, the effective date and effect of that action, and the appeal rights if any; and
(ii) When the certificate is limited, specifies the specialties or subspecialties of tests that the laboratory is no longer authorized to perform, and that are no longer covered under Medicare.
(3) The notice to other entities includes the same information except the information about the laboratory's appeal rights.
(h) Effective date of adverse action.
(1) When the laboratory's deficiencies pose immediate jeopardy, the effective date of the adverse action is at least 5 days after the date of the notice.
(2) When CMS determines that the laboratory's deficiencies do not pose immediate jeopardy, the effective date of the adverse action is at least 15 days after the date of the notice.
[57 FR 7237, Feb. 28, 1992; 57 FR 35761, Aug. 11, 1992, as amended at 68 FR 3714, Jan. 24, 2003]
§ 493.1846 Civil action.
If CMS has reason to believe that continuation of the activities of any laboratory, including a State-exempt laboratory, would constitute a significant hazard to the public health, CMS may bring suit in a U.S. District Court to enjoin continuation of the specific activity that is causing the hazard or to enjoin the continued operation of the laboratory if CMS deems it necessary. Upon proper showing, the court shall issue a temporary injunction or restraining order without bond against continuation of the activity.
§ 493.1850 Laboratory registry.
(a) Once a year CMS makes available to physicians and to the general public specific information (including information provided to CMS by the OIG) that is useful in evaluating the performance of laboratories, including the following:
(1) A list of laboratories that have been convicted, under Federal or State laws relating to fraud and abuse, false billing, or kickbacks.
(2) A list of laboratories that have had their CLIA certificates suspended, limited, or revoked, and the reason for the adverse actions.
(3) A list of persons who have been convicted of violating CLIA requirements, as specified in section 353(1) of the PHS Act, together with the circumstances of each case and the penalties imposed.
(4) A list of laboratories on which alternative sanctions have been imposed, showing—
(i) The effective date of the sanctions;
(ii) The reasons for imposing them;
(iii) Any corrective action taken by the laboratory; and
(iv) If the laboratory has achieved compliance, the verified date of compliance.
(5) A list of laboratories whose accreditation has been withdrawn or revoked and the reasons for the withdrawal or revocation.
(6) All appeals and hearing decisions.
(7) A list of laboratories against which CMS has brought suit under § 493.1846 and the reasons for those actions.
(8) A list of laboratories that have been excluded from participation in Medicare or Medicaid and the reasons for the exclusion.
(b) The laboratory registry is compiled for the calendar year preceding the date the information is made available and includes appropriate explanatory information to aid in the interpretation of the data. It also contains corrections of any erroneous statements or information that appeared in the previous registry.
Subpart S [Reserved]
Subpart T - Consultations
Source:
57 FR 7185, Feb. 28, 1992, unless otherwise noted.
§ 493.2001 Establishment and function of the Clinical Laboratory Improvement Advisory Committee.
(a) HHS will establish a Clinical Laboratory Improvement Advisory Committee to advise and make recommendations on technical and scientific aspects of the provisions of this part 493.
(b) The Clinical Laboratory Improvement Advisory Committee will be comprised of individuals involved in the provision of laboratory services, utilization of laboratory services, development of laboratory testing or methodology, and others as approved by HHS.
(c) HHS will designate specialized subcommittees as necessary.
(d) The Clinical Laboratory Improvement Advisory Committee or any designated subcommittees will meet as needed, but not less than once each year.
(e) The Clinical Laboratory Improvement Advisory Committee or subcommittee, at the request of HHS, will review and make recommendations concerning:
(1) Criteria for categorizing nonwaived testing;
(2) Determination of waived tests;
(3) Personnel standards;
(4) Facility administration and quality systems standards.
(5) Proficiency testing standards;
(6) Applicability to the standards of new technology; and
(7) Other issues relevant to part 493, if requested by HHS.
(f) HHS will be responsible for providing the data and information, as necessary, to the members of the Clinical Laboratory Improvement Advisory Committee.
[57 FR 7185, Feb. 28, 1992, as amended at 58 FR 5237, Jan. 19, 1993; 60 FR 20051, Apr. 24, 1995; 68 FR 3714, Jan. 24, 2003]